青霉素类药物的结构改造
- 格式:ppt
- 大小:324.50 KB
- 文档页数:6

青霉素的分类青霉素是一类广泛应用于临床的抗生素药物,它的发现和应用对医学界产生了革命性的影响。
青霉素的分类是基于其化学结构和抗菌谱的不同特点而进行的,不同类型的青霉素在临床上有着不同的应用范围和疗效。
一、天然青霉素天然青霉素是由真菌产生的抗生素,最早由亚历山大·弗莱明于1928年发现。
这类青霉素的代表性药物是青霉素G,也被称为普鲁卡因青霉素。
天然青霉素具有广谱的抗菌活性,对许多革兰阳性细菌和一些革兰阴性细菌都有较好的抑制作用。
然而,由于其结构较为简单,易被细菌产生的酶类降解,因此其抗菌活性较弱,且易出现耐药性。
二、半合成青霉素半合成青霉素是在天然青霉素的基础上通过化学合成进行改造得到的药物。
通过改变青霉素的侧链结构,可以增强其抗菌活性和稳定性。
半合成青霉素的代表性药物是苄青霉素(青霉素V),也被称为青霉素V钾盐。
苄青霉素对革兰阳性细菌和一些革兰阴性细菌具有较好的抑制作用,且相对于天然青霉素来说更稳定,口服吸收良好。
三、广谱青霉素广谱青霉素是在半合成青霉素的基础上进一步改进得到的药物,具有更广泛的抗菌谱。
其中最重要的代表是氨苄青霉素(青霉素G的衍生物)和阿莫西林(苄青霉素的衍生物)。
广谱青霉素对革兰阳性细菌和革兰阴性细菌均有较好的抑制作用,且对某些耐药菌株仍然有效。
广谱青霉素在临床上广泛应用于治疗呼吸道、泌尿道、皮肤软组织等感染疾病。
四、抗酶青霉素抗酶青霉素是通过改变青霉素分子结构,使其能够抵抗细菌产生的酶类降解而得到的药物。
这类青霉素具有较强的抗菌活性,能够有效抵抗细菌产生的酶类降解,从而提高药物的稳定性和疗效。
抗酶青霉素的代表性药物是氨甲苄青霉素(苄青霉素的衍生物)。
抗酶青霉素在临床上常用于治疗对青霉素敏感但产生酶类抗药性的细菌感染。
五、延长青霉素延长青霉素是通过在青霉素分子结构中引入特殊的化学基团,从而延长药物在体内的半衰期,减少用药频率,提高疗效的药物。
这类青霉素的代表性药物是苄唑青霉素和氨苄唑青霉素。

高纲1038省高等教育自学考试大纲01759药物化学〔二〕医科大学编省高等教育自学考试委员会办公室一、课程性质与其设置目的与要求〔一〕课程性质和特点《药物化学》是高等教育自学考试药学专业的一门专业根底课,本课程重点论述药物的化学结构、命名合、制备原理、理化性质和构效关系。
通过学习可以使考生对常用药物的制备原理、理化性质、构效关系与其应用有系统的认识,达到熟悉并理解合理有效地使用常用化学药物这一目的。
本课程的目标是使学生通过本课程的学习,掌握现代药物化学根本理论和技能,对常用药物的结构类型、制备原理、理化性质和构效关系、与其应用应用有一个较系统的认识,并了解现代药物化学的开展,为以后的学习与在医药工作实践中理有效的使用常用药物打下坚实根底。
〔二〕本课程的根本要求通过本课程的学习,应达到如下要求:1.掌握常用药物的名称、化学名、化学结构、理化性质、用途与其中一些药物的合成方法,掌握重要药物类型的构效关系。
2.掌握药物在贮存过程中可能发生的化学变化与其化学结构和稳定性之间的关系,以保证用药的安全、有效。
3.掌握以光学活性体供药的药物的立体化学结构、生物活性特点。
4.掌握新药发现与研究根本的原理和方法。
〔三〕本课程与相关课程的关系本课程的前修课程是有机化学,无机化学,分析化学以与药理学等。
二、课程容与考核要点第一章绪论一、课程容本章主要介绍了药物化学研究的起源和开展以与药物的命名。
二、学习目的与要求通过本章的学习,掌握新药研究与开发的容,掌握常见药物的命名方法。
三、考核知识点与考核要求1.领会药物化学的起源和开展,药物的命名方法。
2.掌握药物化学的概念、研究容、药物命名原那么。
3.熟练掌握常见的药物作用的靶点, INN采用的常见药物的词干的中文译名。
第二章中枢神经系统药物一、课程容本章主要介绍了镇静催眠药、抗癫痫药、抗精神失常药、抗抑郁药、镇痛药、中枢兴奋药的作用机制、结构特征和与其典型药物的化学结构、理化性质、用途、代与其中一些药物的合成方法,构效关系。

青霉素结构的探究摘要青霉素是人类抗菌历史上最伟大的产物。
在极其简陋的实验条件下,正是由于科学家不懈地探索,青霉素神秘的结构才逐渐展现在人类面前。
现在广泛用于临床上的β-内酰胺抗生素,大都是在青霉素原有结构基础上修饰改造而来。
关键词青霉素立体构型结构改造青霉素(Penicillin),音译名盘尼西林,人类历史上最负盛名的抗生素,它的研制成功大大增强了人类抵抗细菌感染的能力,带动了抗生素家族的诞生。
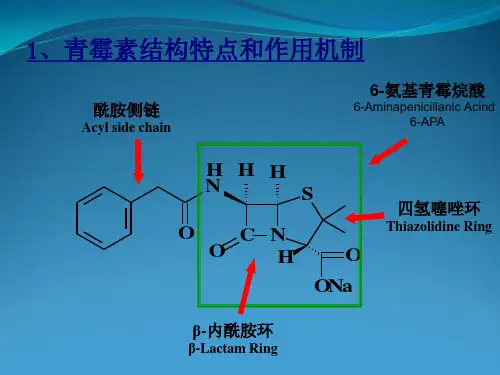
由于分子中含有4个原子构成的β-内酰胺结构(图1),故统称为β-内酰胺抗生素。
青霉素分子由氢化噻唑环与β-内酰胺环并和而成,二者构成青霉素分子的母核,在母核上分别连有羧基和酰氨基侧链。
β-内酰胺环为一个平面结构,但2个稠和环不共平面。
青霉素分子中含有3个手性碳原子,只有3个碳原子绝对构型为2S,5R,6R的具有抗菌活性。
从青霉菌培养液中得到6种天然青霉素,现已证实为侧链不同的青霉素(见表1)。
其中以青霉素G的含量最高,效用最好,故在临床上广泛使用。
1 青霉素结构的探索对青霉素结构工作的探索是极其曲折的。
在那个设备粗糙、条件简陋的年代,科学家对青霉素研究的困难程度是现代科学家所无法想象的。
1.1 分子式的确定早期实验曾指出青霉素分子中不含S原子,这个错误的结论直到1943年7月才被纠正。
不同的青霉素水解都可以得到一种氨基酸——青霉胺,其分子式是C5H11NO2S,除此之外还有不同的青霉醛和二氧化碳。
从反应的产物可看出,青霉素分子中含有2个氮原子,4个氧原子和1个硫原子。
再后来研究发现2-戊烯基青霉素的钠盐分子式为C14H19N2O4SNa,苄基青霉素的钠盐分子式为C16H17N2O4SNa。
1.2 6种不同的青霉素化学家们在刚着手研究青霉素时就遇到了很大的困难,在自然界中不止存在一种天然的青霉素。
在英国,采用弗莱明发现青霉素时的表面培养法获得的青霉素与在美国采用玉米浸渍液培养出来的青霉素不一致,后来又陆续发现了另外一些共6种天然的青霉素(表1)。

01759药物化学(二)简答题1、先导化合物进行前药修饰的目的是什么?(1)增加脂溶性以提高吸收性能;(2)部位特异性;(3)增加药物的化学稳定性:(4)消除不适宜的制剂性质;(5)延长作用时间。
2、简述发现先导物的主要途径。
(1)由天然有效成分获得,包括植物、微生物和内源性活性物质;(2)反义核普酸;(3)基于生物大分子结构和作用机理设计;(4)组合化学;(5)基于生物转化发现。
3、利用前药原理对药物进行结构修饰,可以改变药物的哪些性质?(1)提高药物的组织选择性;(2)提高药物的稳定性;(3)延长药物作用时间;(4)改善药物的吸收;(5)改善药物的溶解度;(6)消除药物的不良味觉;(7)发挥药物的配伍作用。
4、前药的主要特征是什么?(1)原药与暂时转运基团以共价键连接,并且在体内可断裂,形成原药;(2)前药无活性或活性低于原药;(3)前药与暂时转运基团无毒性;(4)前药在体内产生原药的速率是快速的,以保障原药在作用部位有足够的药物浓度,并且应尽量减低前药的直接代谢。
5、叙述前药与软药设计的区别。
(1)前药是指用化学方法由有活性原药转变的无活性衍生物,后者在体内经酶或非酶解作用释放出原药而发挥疗效。
(2)软药系本身其有活性,在体内产生药理作用后可按预知方式和可控速率经进一步代谢转化成无活性产物的药物。
6、药物的第Ⅰ相生物转化的主要目的是什么?第II相生物转化的主要途径有哪几种?第Ⅰ相生物转化的主要目的是增加药物的极性,使之容易排泄。
第Ⅰ相生物转化有如下几种途径①葡萄糖醛酸结合;②硫酸结合;③氨基酸结合;④谷胱甘肽或疏基尿酸结合;⑤甲基化反应;⑥乙酰化反应。
7、简述吗啡及合成镇痛药的立体结构特征。
(1)分子中具有一个平坦的芳环结构,与受体的平坦区通过范德华力结合;(2)有一个叔氮原子的碱性中心,在生理pH条件下,大部分电离为阳离子正电中心,与受体表面的阴离子部位缔合;(3)联结它们两者之间的烃链部分突出于平面的前方,正好与受体的凹槽相适应。





碳青霉烯类碳青霉烯类是抗菌谱最广,抗菌活性最强的非典型β-内酰胺抗生素,已成为治疗严重细菌感染最主要的抗菌药物之一。
主要药物有:亚胺培南、美洛培南、帕尼培南、比阿培南结构:碳青霉烯类抗生素是由青霉素结构改造而成的一类新型β-内酰胺类抗生素,问世于20世纪80年代。
其结构与青霉素类的青霉环相似,不同之处在于噻唑环上的硫原子为碳所替代,且C2与C3之间存在不饱和双键;另外,其6位羟乙基侧链为反式构象。
研究证明,正是这个构型特殊的基团,使该类化合物与通常青霉烯的顺式构象显著不同,具有超广谱的、极强的抗菌活性,以及对β-内酰胺酶高度的稳定性。
抗菌谱和抗菌活性:对大多数革兰阳性、阴性需氧菌、厌氧菌及多重耐药菌均有较强的抗菌活性,但MRSA、屎肠球菌、嗜麦芽窄食单胞菌等对本品天然耐药。
美洛培南对葡萄球菌和肠球菌的作用较亚胺培南弱2-4倍,对耐甲氧西林葡萄球菌、屎肠球菌同样耐药;但对肠杆菌科细菌的抗菌活性是亚胺培南的2-16倍,对铜绿假单胞菌的抗菌活性是亚胺培南的2-4倍。
帕尼培南对G+菌的抗菌活性与亚胺培南相仿或略强,对肠杆菌科细菌的抗菌活性与亚胺培南相仿,对铜绿假单胞菌的抗菌活性则逊于亚胺培南。
比阿培南对肠道杆菌科和绿脓杆菌的抗菌活性比亚胺培南强,对葡萄球菌和肠球菌较亚胺培南弱。
对β-内酰胺酶的稳定性:碳青霉烯类对质粒介导的超广谱β-内酰胺酶(ESBLs)、染色体及质粒介导的头孢菌素酶(AmpC酶)均具有高度稳定性。
但可被金属β-内酰胺酶水解灭活,造成碳青酶烯类抗生素耐药。
作用机制:碳青霉烯类抗生素作用方式都是抑制胞壁粘肽合成酶,即青霉素结合蛋白(PBPs),从而阻碍细胞壁粘肽合成,使细菌胞壁缺损,菌体膨胀致使细菌胞浆渗透压改变和细胞溶解而杀灭细菌。
耐药机制:主要有:1.青霉素结合蛋白结合力下降,主要见于MRSA及某些肠球菌;2.Ⅰ型β-内酰胺酶水解碳青霉烯类的微弱活性加上细菌对碳青霉烯类通透性下降致耐药性产生,主要见于一些肠杆菌及绿脓杆菌;3.菌株产生含锌β-内酰胺酶水解碳青霉烯类,这些菌株大多为临床非常见的病原菌。

1.天然青霉素G有哪些缺点?试述半合成青霉素的结构改造方法。
答:天然青霉素G的缺点为对酸不稳定,不能口服,只能注射给药;抗菌谱比较狭窄,仅对革兰阳性菌的效果好;细菌易对其产生耐药性;有严重的过敏性反应。
在青霉素的侧链上引入吸电子基团,阻止侧链羰基电子向β一内酰胺环的转移,增加了对酸的稳定性,得到一系列耐酸青霉素。
在青霉素的侧链上引入较大体积的基团,阻止了化合物与酶活性中心的结合。
又由于空间阻碍限制酰胺侧链R与羧基间的单键旋转,从而降低了青霉素分子与酶活性中心作用的适应性,因此药物对酶的稳定性增加。
在青霉素的侧链上引入亲水性的基团(如氨基,羧基或磺酸基等),扩大了抗菌谱,不仅对革兰阳性菌有效,对多数革兰阴性菌也有效。
2.简述现代新药开发与研究的内容。
3.巴比妥类药物的一般合成方法中,用卤烃取代丙二酸二乙酯的氢时,当两个取代基大小不同时,一般应先引入大基团,还是小基团?为什么?答:当引入的两个烃基不同时,一般先引入较大的烃基到次甲基上。
经分馏纯化后,再引入小基团。
这是因为,当引入一个大基团后,因空间位阻较大,不易再接连上第二个基团,成为反应副产物。
同时当引入一个大基团后,原料、一取代产物和二取代副产物的理化性质差异较大,也便于分离纯化。
4.以captopril为例,简要说明ACEI类抗高血压药的作用机制及为克服captopril的缺点及对其进行结构改造的方法。
答:血管紧张素转化酶抑制剂(ACEI)类抗高血压药主要是通过抑制血管紧张素转化酶(ACE)的活性、,使血管紧张素I(AngI)不能转化为血管紧张素Ⅱ(AngⅡ),导致血浆中AngⅡ数量下降,无法发挥其收缩血管的作用及促进醛固酮分泌作用,ACEI还能抑制缓激肽的降解,上述这些作用结果均使血压下降。
卡托普利(Captopril)是根据ACE的结构设计出来的第一个上市的ACEI,为脯氨酸的衍生物,脯氨酸氮原子上连一个有甲基和巯基取代的丙酰基侧链,使Captopril具有良好的抗高血压作用,但用药后易产生皮疹、干咳、嗜酸性粒细胞增高、味觉丧失和蛋白尿的副作用.,味觉丧失可能与结构中的巯基有关,考虑到脯氨酸的吡咯环及环上的羧基阴离子对结合酶部位起到重要的作用,故在尽可能保留该部分结构特点的同时,用α一羧基苯丙胺代替巯基如依那普利(Enalapril),或用含次膦酸基的苯丁基代替巯基福辛普利(Fosinpril),再将羧基或次膦酸基成酯,则可得到一类长效的ACEI,上述不良反应也减少。
半合成青霉素的结构改造方法引言半合成青霉素是一种重要的抗生素,由于其广谱抗菌活性和良好的耐受性,被广泛应用于临床医学领域。
然而,青霉素的天然产物含量较低,大规模生产困难,因此,寻找半合成方法来改造青霉素的结构以提高其产量成为一个重要的研究方向。
本文将详细探讨半合成青霉素的结构改造方法。
I. 青霉素的结构与合成方法回顾1. 青霉素的结构青霉素是一种含有β-内酰胺环的混合酸氨酯类抗生素,其结构由苯丙侧链、β-内酰胺环和五元内酯环组成。
2. 青霉素的天然合成路径青霉素的天然合成路径包括青霉素酸的生物合成和β-内酰胺环、五元内酯环的合成。
青霉素酸由L-α-酸乳酸经过环化反应形成β-内酰胺环,然后与半胱氨酸形成五元内酯环。
3. 青霉素的全合成方法青霉素的全合成方法较为复杂,包括多步反应和复杂条件,因此不适合用于大规模生产。
为了解决这一问题,研究人员提出了半合成方法来改造青霉素的结构以提高产量。
II. 基于半合成的青霉素结构改造方法1. 引入新的化学修饰基团通过在青霉素分子中引入新的化学修饰基团,可以改变其物理化学性质和抗菌活性。
例如,引入氯原子可以增强青霉素的抗菌活性。
这种方法需要合成新的衍生物,并通过活性筛选来评估其抗菌活性。
2. 修改侧链结构通过修改苯丙侧链结构,可以改变青霉素的溶解性、稳定性和生物利用度。
例如,引入疏水基团可以提高青霉素在脂肪组织中的分配,从而增强其抗菌活性。
3. 调节β-内酰胺环和五元内酯环的形成通过调节β-内酰胺环和五元内酯环的形成,可以改变青霉素的稳定性和抗菌活性。
例如,通过改变环化反应的反应条件和催化剂,可以调节β-内酰胺环的形成速度和稳定性。
4. 制备半合成前体物制备半合成前体物是半合成青霉素的关键步骤。
通过合成不同的前体物,可以改变青霉素的结构和物理化学性质。
例如,通过合成β-内酰胺环和五元内酯环的前体物,可以改变青霉素的稳定性和活性。
III. 半合成青霉素的应用与展望半合成青霉素在临床医学领域具有广阔的应用前景。
青霉素类药物的缺点及其结构修饰原理及方法
缺点:青霉素的价格是比较便宜,但是疗效是不错的。
现在很多的新药都是从青霉素衍生而来。
现在医院里面使用的量较少,是因为很多头孢类药物的使用,导致了细菌对青霉素耐药或者疗效打了折扣。
所以医生使用青霉素的时候,如果效果一般,就会及时更换药物。
还是青霉素是可以进入大脑,治疗脑膜炎的一种,目前依然在使用的一种有效抗生素。
但是青霉素的缺点也是有很多的,例如最严重的过敏反应,轻者可以引起水肿、皮肤红等,重的话可以引起休克、或者是死亡的。
所以使用之前一定要皮试的,阳性者不能用。
然后就是容易使细菌产生耐药性,这是现在普遍存在的问题的了。
结构作用方法:青霉素之所以具有强大的抗菌作用是由于青霉素与细菌细胞壁可以发生作用。
黏肽是细菌细胞壁的主要成分,也是细菌细胞壁中最坚硬的一层。
它的存在可以维持细菌细胞的外形,保持其细胞壁的通透性。
青霉素的结构同黏肽的末端结构丙氨酰丙氨酸相似,其可以取代丙氨酰丙氨酸与酶的活性中心结合,从而使组成黏肽的多肽不能交联形成网状的黏肽,导致细菌细胞壁不能形成,从而使细菌被溶解而死亡。
而人类的细胞没有细胞壁,只有细胞膜,所以人类细胞受青霉素的影响很小。
青霉素的合成方法与工艺优化策略青霉素是一种广泛应用于临床治疗的抗生素,被誉为“抗生素之王”。
它是由青霉菌属真菌产生的一类天然产物,具有广谱的抗菌活性,对革兰阳性细菌尤为有效。
本文将探讨青霉素的合成方法以及工艺优化策略,以期为医学人员提供更多的科学依据和实践指导。
一、青霉素的合成方法青霉素的合成方法主要分为天然合成和半合成两种。
1. 天然合成:青霉素的天然合成是通过青霉菌属真菌自身的代谢途径合成的。
青霉素的合成过程包括青霉素酸的合成、侧链的合成以及酸酐的合成等。
其中,青霉素酸是青霉素的前体,通过一系列酶的作用,最终合成出青霉素。
2. 半合成:半合成是在天然合成的基础上,通过化学手段对青霉素的结构进行改造,以获得更多种类和更高效的青霉素类似物。
半合成青霉素的合成方法主要包括侧链改造、半合成酸酐和半合成青霉素的合成等。
二、青霉素的工艺优化策略青霉素的工艺优化策略主要包括改进合成方法、提高产量和纯度、减少污染物产生等方面。
1. 改进合成方法:通过改进合成方法,可以提高青霉素的产量和纯度,并减少副产物的生成。
例如,引入新的催化剂、优化反应条件、改变反应顺序等,可以提高合成效率和产物纯度。
2. 提高产量和纯度:青霉素的产量和纯度是评价合成工艺的重要指标。
通过优化培养条件、改进发酵工艺、提高菌株的发酵能力等手段,可以提高青霉素的产量和纯度。
3. 减少污染物产生:在青霉素的合成过程中,会产生一些副产物和污染物,对产品质量和纯度产生不利影响。
通过优化反应条件、改进分离纯化工艺、加强废水处理等措施,可以减少污染物的产生,提高产品质量。
4. 提高抗菌活性:除了改进合成方法和工艺优化,还可以通过改变青霉素的结构,提高其抗菌活性。
例如,通过半合成的方法,可以引入新的官能团或改变侧链结构,以增强青霉素的抗菌活性。
总结:青霉素的合成方法和工艺优化策略是医学领域的重要研究方向。
通过不断改进合成方法、提高产量和纯度、减少污染物产生等措施,可以获得更高效、更纯净的青霉素产品,为临床治疗提供更好的药物选择。
药化思考题(2)第二章抗肿瘤药1、按作用机理,抗肿瘤药药物的分类。
各类的结构类型、作用机理及代表药物。
作用机理:(1)直接作用于DNA,破坏其结构和功能的药物;(2)干扰DNA和核酸合成的药物;(3)抗有丝分裂,影响蛋白质合成的药物;(4)作用于肿瘤信号转导机制的药物一、直接作用于DNA的药物1、烷化剂:(定义)(1)氮芥类:脂肪氮芥(盐酸氮芥);芳香氮芥(苯丁酸氮芥);氨基酸氮芥(美法伦、氮甲)杂环氮芥(环磷酰胺,又名癌得星)作用机制:在DNA鸟嘌呤间进行交链结合,阻断DNA的复制(2)乙撑亚胺类:曲他胺(TEM)、替哌(TEPA)、塞替哌、丝裂霉素C(3)甲磺酸脂类:白消安(4)亚硝基脲类:卡莫司汀2、金属铂配合物:顺铂、卡铂作用机理:活泼离子与DNA分子两个嘌呤碱基洛合形成螯合物,嵌在DNA上,使DNA断键,顺式与DNA作用,反式无效。
3、直接作用于DNA的天然产物:博来霉素(BLM)4、DNA拓扑酶抑制剂:(1)Topo I抑制剂(作用于单链):喜树碱、拓扑替康、(2)Topo Ⅱ抑制剂(作用于双链):多柔比星、阿霉素、柔红霉素二、干扰DNA合成和核酸的合成(抗代谢肿瘤的药物)1、嘧啶拮抗物(1)尿嘧啶衍生物:氟尿嘧啶(5-Fu)、(2)胞嘧啶衍生物:盐酸阿糖胞苷(APA-C)2、嘌呤拮抗物:巯嘌呤(6-MP)3、叶酸拮抗物:甲氨蝶呤(MTX)、氨基蝶呤三、抗有丝分裂的药物:秋水仙碱、长春碱类、紫杉醇2、重点掌握烷化剂类和干扰DNA合成类(代谢拮抗)3、重点药物:环磷酰胺、顺铂、氟尿嘧啶、阿糖胞苷、多柔比星、巯嘌呤、甲氨蝶呤4、了解药物:丝裂霉素C、博来霉素、紫杉醇第三章抗病毒药和抗艾滋病药1、了解抗病毒药物的主要靶点和各类药物的结构类型抗病毒药的靶点(作用方式)1、阻止病毒在细胞上吸附:丙种球蛋白2、阻止病毒穿入细胞:金刚烷胺类3、阻止病毒复制核酸:核苷类4、增强免疫:干扰素2、干扰病毒核酸复制的药物3、掌握核苷类抗病毒药物基本结构及设计原理(代谢拮抗)合成过程中,竞争性地抑地DNA聚合酶或RNA聚合酶,从而抑制酶的活性,干扰病毒核酸的合成,产生抗病毒的作用。