P53在凋亡中的信号通路 PPT 可修改
- 格式:ppt
- 大小:306.00 KB
- 文档页数:1

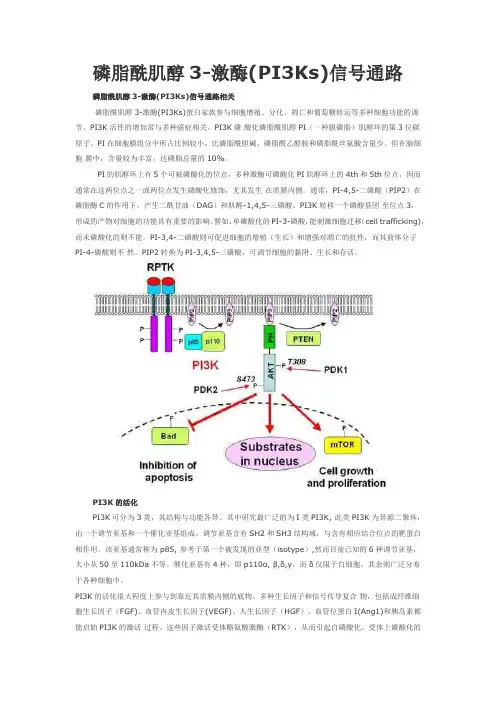
磷脂酰肌醇3-激酶(PI3Ks)信号通路磷脂酰肌醇3-激酶(PI3Ks)信号通路相关磷脂酰肌醇3-激酶(PI3Ks)蛋白家族参与细胞增殖、分化、凋亡和葡萄糖转运等多种细胞功能的调节。
PI3K活性的增加常与多种癌症相关。
PI3K磷酸化磷脂酰肌醇PI(一种膜磷脂)肌醇环的第3位碳原子。
PI在细胞膜组分中所占比例较小,比磷脂酰胆碱、磷脂酰乙醇胺和磷脂酰丝氨酸含量少。
但在脑细胞膜中,含量较为丰富,达磷脂总量的10%。
PI的肌醇环上有5个可被磷酸化的位点,多种激酶可磷酸化PI肌醇环上的4th和5th位点,因而通常在这两位点之一或两位点发生磷酸化修饰,尤其发生在质膜内侧。
通常,PI-4,5-二磷酸(PIP2)在磷脂酶C的作用下,产生二酰甘油(DAG)和肌醇-1,4,5-三磷酸。
PI3K转移一个磷酸基团至位点3,形成的产物对细胞的功能具有重要的影响。
譬如,单磷酸化的PI-3-磷酸,能刺激细胞迁移(cell trafficking),而未磷酸化的则不能。
PI-3,4-二磷酸则可促进细胞的增殖(生长)和增强对凋亡的抗性,而其前体分子PI-4-磷酸则不然。
PIP2转换为PI-3,4,5-三磷酸,可调节细胞的黏附、生长和存活。
PI3K的活化PI3K可分为3类,其结构与功能各异。
其中研究最广泛的为I类PI3K, 此类PI3K为异源二聚体,由一个调节亚基和一个催化亚基组成。
调节亚基含有SH2和SH3结构域,与含有相应结合位点的靶蛋白相作用。
该亚基通常称为 p85, 参考于第一个被发现的亚型(isotype),然而目前已知的6种调节亚基,大小从50至110kDa不等。
催化亚基有4种,即p110α, β,δ,γ,而δ仅限于白细胞,其余则广泛分布于各种细胞中。
PI3K的活化很大程度上参与到靠近其质膜内侧的底物。
多种生长因子和信号传导复合物,包括成纤维细胞生长因子(FGF)、血管内皮生长因子(VEGF)、人生长因子(HGF)、血管位蛋白I(Ang1)和胰岛素都能启始PI3K的激活过程。



幻21) p53研究30年2)p53的定义3)p53基因的结构与表达4)p53蛋白的功能5)p53的信号通路6)p53的基因治疗幻3p53研究30年1979年,英国癌症研究基金会、美国普林斯顿大学的研究者首次追踪到了p53基因的踪迹。
最初,p53基因并未受到重视,甚至在最初的10年中,p53一直被视为能够诱发肿瘤产生的癌基因。
众所周知,一条基因由一系列脱氧核糖核酸按照相应的顺序彼此串联而成,如果其中的某个或某些核苷酸发生改变就意味着这条基因发生了突变,而起初研究者拿到的基因就是p53的突变版本,按照这一版本翻译成的蛋白质自然就无法行使正常p53基因的功能。
十年之后,约翰霍普金斯医学院的研究者终于找到了正确的p53基因,即野生型p53。
不但如此,科学家的发现还为这一基因摘掉了癌基因的恶名:与此前认识恰恰相反的是,p53是一个在人体内发挥广泛作用的强有力的抑癌基因。
p53在众多信号通路中起着枢纽的作用,调节着很多非常重要的生物学活性,包括维持基因组稳定、诱导细胞凋亡等等。
有研究发现p53是可选择性剪接的基因家族中的一员。
突变的p53基因具有某些特性,它能够对肿瘤的发生、发展和转移进行控制。
对p53的认识一直在不断加深,同时p53的治疗作用也早已进入临床应用,现在很多癌症治疗方案都试图重建其在癌基因中的表达。
幻4p53定义但是P53基因突变后,由于其空间构象发生改变,失去了对细胞生长、凋亡和DNA 修复的调控作用,P53 基因由抑癌基因转变为癌基因。
幻5p53基因的结构正常的P53蛋白在细胞中易水解,野生型p53蛋白的半衰期很短,因此用免疫组化法检测不到野生型p53蛋白,而突变型p53蛋白降解缓慢,代谢半衰期比较长,积聚在核内易于检测到,所以应用免疫组化检测的p53均为突变型,可作为多种癌的基因标志。
P53蛋白N 端有一个与转录因子相似的酸性结构域,与GAL4(GAL4是酵母转录激活蛋白Gal4的基因)的DNA结合区重组时,融合蛋白能激活GAL4操纵子转录,激活功能定位在P53第20~40位密码子。


p53 SignalingRT² Profiler™ PCR Arrayp53 Signaling Pathway PCR ArrayCellular Senescence PCR ArrayDNA Damage Signaling Pathway PCR ArrayCell Cycle PCR ArraySureSilencing RNAip53 Signaling Pathway Gene RNAiCellular Senescence Gene RNAiDNA Damage Signaling Pathway Gene RNAiCell Cycle Gene RNAiCignal™ Reporter Assaysp53 Pathway Reporter Assay KitE2F Reporter Assay KitEGR1 Reporter Kitp53 is a tumour suppressor protein that regulates the expression of a wide variety of genes involved in Apoptosis, Growth arrest, Inhibition of cell cycle progression, Differentiation and accelerated DNA repair or Senescence in response to Genotoxic or Cellular Stress. As a transcription factor, p53 is compos ed of an N-terminal Activation Domain, a central specific DNA Binding Domain, and a C-terminal Tetramerization Domain, followed by a Regulatory Domain rich in basic Amino acids. Having a short half-life, p53 is normally maintained at low levels in unstress ed mammalian cells by continuous ubiquitylation and subsequentdegradation by the 26S Proteasome. Nonphosphorylated p53 is ubiquitylated by the MDM2 (Mouse Double Minute-2) ubiquitin ligase. MDM2 binding inactivates p53 by two mechanisms. First, MDM2 binds to the transactivation domain of p53, precluding interaction with the transcriptional machinery. Second, this binding mediates the covalent attachment of ubiquitin to p53. Ubiquitylated p53 is then degraded by the Proteasome. Thus MDM2 acts as a major reg ulator of the tumor suppressor p53 by targeting its destruction. When the cell is confronted with stress like DNA damage, Hypoxia, Cytokines, Metabolic changes, Vi ral infection, or Oncogenes, however, p53 ubiquitylation is suppressed and p53 accumulates in the nucleus, where it is activated and stabilized by undergoing multiple covalent modifications including Phosphorylation and Acetylation (Ref.1 & 2).Phosphorylation of p53 mostly occurs in the N-terminal activation domain at the Ser6, Ser9, Ser15, Thr18, Ser20, Ser33, Ser37, Ser46, Thr55, and Thr81 residues, with some phosphorylation occurring in the C-terminal linker and basic regions at Ser315, Ser371, Ser376, Ser378, and Ser392. Phosphorylation on most of these sites is induced by DNA damage, with som e, such as Thr55 and Ser376, being repressed upon genotoxic stress. p53 phosphorylation is mediated by several cellular kinases including Chks (Checkpoint Kinases), CSNK1-Delta (Casein Kinase-1-Delta), CSNK2 (Casein Kinase-2), PKA (Protein Kinase A), CDK7 (Cyclin-Dependent Kinase-7), DNA-PK (DNA-Activated- Protein Kinase), HIPK2 (Homeodomain-Interacting Protein Kinase-2), CAK (CDK-Activating Kinase), p38 and JNK (Jun NH2-terminal kinase). Notably, phosphorylation at Ser15 by ATM (Ataxia Telangiectasia Mutated Gene)/ATR (Ataxia-Telangiectasia and Rad3 Related), either directly or through Chk1 (Cell Cycle Checkpoint Kinase-1)/Chk2 (Cell Cycle Checkpoint Kinase-2), or at Ser20 by Chk1/Chk2 has been shown to alleviate the inhibition or degradation of p53, leading to p53 stabilization and activation. The phosphorylation-induced p53 stabilization and activation are mediated through multiple mechanisms and may vary according to the cellular context or microenvironment. HIF-1Alpha (Hypoxia-Inducible Factor-1-Alpha) has been implicated to be involved in p53 stabilization, the precise mechanism by which HIF-1Alpha regulates p53-mediated function remains unknown. Recently, the interaction between p53 and HIF-1Alpha was reported to evoke HIF-1Alpha degradation. Members of the PIAS (Protein Inhibitor of Activated STAT) protein family have also been found to interact with p53. PIAS1 and PIAS-Gamma function as SUMO (Small Ubiquitin Related Modifier-1) ligases for p53. Moreover, the RING finger domain of PIAS1 binds to the C-terminus of the tumor suppressor p53 and catalyzes its sumoylation, a modification which represses p53 activity on a reporter plasmid containing consensus p53 DNA binding sites. PML (Promyelocytic Leukemia) also activates p53 by recruiting it to multiprotein complexes termed PML-nuclear bodies. PML is a tumor suppressor protein and the major component of multiprotein nuclear complexes that have been variably termed Kremer bodies, ND10, PODs (for PML Oncogenic Domains), and PML-NBs (PML-Nuclear Bodies). PML binds directly with p53 and recruits it to PML-NBs. Recruitment to PML-NBs activate p53 by bringing it in close proximity with CBP (CREB-Binding Protein) /p300. BRCA1 (Breast Cancer-1 Gene) and p53 can also physically associate, both in vitro and in vivo and function in a common pathway of tumor suppression. The ability of BRCA1 to biochemically modulate p53 function suggests that this may be afundamental role of BRCA1 in tumor suppression (Ref.3, 4 & 5).Another important modification of p53 is acetylation. p53 is specifically acetylated at Lys370, Lys372, Lys373, Lys381, and Lys382 by p300/CBP and at Lys320 by PCAF (p300/CBP-associated factor). Acetylation has been shown to augment p53 DNA binding, and to stimulate p53-mediated transactivation of its downstream target genes through the recruitment of coactivators. Acetylation may also regulate the stability of p53 by inhibiting its ubiquitination by MDM2. In vivo, acetylation at Lys320, Lys373, and Lys382 is induced by many genotoxic agents, including UV-radiation, IR (Ionizing Radiation), hypoxia, oxidative stress, and even depletion of ribonucleotide pools. p53 can also be deacetylated by HDAC1 (Histone Deacetylase-1) and SIRT1. Human SIRT1 is an enzyme that deacetylates the p53 tumor suppressor protein and has been suggested to modulate p53-dependent functions including DNA damage-induced cell death. p53 deacetylation has been suggested to down-regulate the activation of genes such as Bax and p21WAF1. Phosphorylation and acetylation are interdependent. In deed, phosphorylation at the p53N-terminus has been shown to enhance its interaction with acetylase p300/CBP and to potentiate p53 acetylation. Activated p53 functions effectively as a transcription factor and induces transcription of several genes. The D NA targets of p53 are consensussequences consisting of a 10-base pair repeat of 5'-PuPuPu-C(A/T)(T/A)GPyPyPy-3' (where Pu is a purine and Py is a pyrimidine). It also can bind to a palindromic site having a four or five-base pair inverted repeat of a similar sequence. Complete p53 is inactive for specific DNA binding unless activated by covalent and noncovalent modifications of the basic C-terminal domain. After p53 is activated it can be involved in cell-cycle inhibition, apoptosis, genetic repair, and inhibition of blood-vesselformation (Ref.5, 6 & 7).Cell cycle inhibition takes place when there is a block in cell-cycle division. p53 does this by stimulating the expression of p21 WAF1/CIP1 (Cyclin Dependent Kinase Inhibitor-p21). This protein is an inhibitor of CDKs (Cyclin-Dependent Kinases) that regulate the cell cycle via perturbation of their partner cyclin. Cyclins are involved to ensure successful transitions from S phase to G1. Since p21 WAF1/CIP1 inhibits CDKs it results in inhibition of both G1-to-S and G2-to-mitosis transitions by causing hypophosphorylation of Rb (Retinoblastoma) and preventing the release of E2F. Additionally p53 can stimulate 14-3-3, a protein that sequesters Cyclin B1-CDK1 complexes out of the nucleus. This results in a G2 block. Activated p53 may also initiate apoptosis and stop cell proliferation. p53 stimulates a wide network of signals that act through two major apoptotic pathways: Extrinsic Pathways and Intrinsic Pathways. The extrinsic pathway involves engagement of pa rticular `death' receptors that belong to the TNFR (Tumor Necrosis Factor Receptor) family and, through the formation of the DISC(Death-Inducing-Signaling-Complex), leads to a cascade of activation of Caspases, including Caspase8 and Caspase3, which in turn induce apoptosis. Most common death receptors involved in extrinsic apoptosis Fas, DR5 (Death Receptor-5) and PERP. The intrinsic apoptotic pathway is dominated by the Bcl2 (B-Cell CLL/Lymphoma-2) family of proteins, which governs the release of CytoC (Cytochrome-C) from the mitochondria. The Bcl2 family comprises anti-apoptotic (pro-survival) and pro-apoptotic members. The Bcl2 family is divisible into three classes: pro-survival proteins, whose members are most structurally similar to Bcl2, such as BclXL; pro-apoptotic proteins, BAX (Bcl2 Associated-X Protein) and BAK (Bcl2 Antagonist Killer-1), which are structurally similar to Bcl2 and BclXL and antagonize their pro-survival functions; and the pro-apoptotic `BH3-only' proteins. Intriguingly, a key subset of the Bcl2 family genes are p53 targets, including BAX, Noxa, PUMA (p53-Upregulated Modulator of Apoptosis) and the most recently identified, BID (BH3 Interacting Domain Death Agonist). p53 may also inhibit Bcl2 that is an inhibitor of apoptosis. p53 may also have a role in maintaining genetic stability by 'nucleotide-excision' repair of DNA, chromosomal recombination and chromosome segmentation. GADD45 (Growth Arrest- and DNA Damage-Inducible Gene-45) is a multifunctional protein that is regulated by p53 and that may play a role in DNA repair and cell cycle checkpoints. p53 can playa role in the inhibition of blood-vessel formation. In order for tumours to reach a large size, they must initiate the growth of nutrient-bringing blood vessels in their vicinity, the process of angiogenesis. p53 stimulates the production of genes that prevent this process from happening. p53 activates the expression of the Tsp1 (Thrombospondin-1), an anti-angiogenic factor, along with other angiogenesis inhibitor BAI1 (Brain-specific Angiogenesis Inhibitor-1) (Ref. 8, 9 & 10).In addition, p53 regulates MDM2 function in a negative feedback loop, because the MDM2 gene is a target for p53. Therefore, activation of p53 eventually leads to its own inactivation by switching on a pathway that leads to its destruction. MDM2 is subject to further regulation by direct binding of the ARF (Active Response Factor) protein, which prevents MDM2-mediated p53 proteolysis. PTEN (Phosphatase and Tensin Homolog), on the other hand inhibits MDM2-mediated p53 degradation. p53 can transcriptionally activate PTEN, which may further inhibit Akt activity. Therefore, inhibition of Akt by the inhibitors may trigger a positive feedback with perhaps additional anti-tumor effects. The c-Fos proto-oncogene is also a target for transactivation by the p53 tumor suppressor. Mutations in p53 are associated with genomic instability and increased susceptibility to cancer. It is the most frequently mutated protein in all cancer with an estimated 60% of all cancers having mutated forms that affect its growth suppressing activities. However some common tumours have a higher incidence such that 90% of cervical and 70% of colorectal are found to have p53 mutations. The p53 protein can be inactivated in several ways, in cluding inherited mutations that result in a higher incidence of certain familial cancers such as Li-Fraumeni syndrome. Certain DNA tumour viruses, such as the human adenovirus and the papilomavirus, bind to and inactivate the protein. Functional p53 is th ought to provide a protective effectagainst tumorigenesis (Ref.2, 11 & 12).。
