PCR扩增16SrDNA
- 格式:pdf
- 大小:1.07 MB
- 文档页数:22

小量提取细菌基因组总DNA①挑取LB平板上新鲜活化的单菌落于5mlLB液体培养基中,35-37℃震荡过夜培养;②转移3mL菌液于5mL离心管中,4℃,12000rpm,离心5min,弃掉上清;③将细菌沉淀物用0.5mol/l的NaCl洗一次,尽量空干上清;(此步可省略)④将沉淀重悬于1mL 50mmol/l的Tris(pH 8.0)缓冲液中,然后加入0.2mL 新鲜配置的溶菌酶溶液(10mg/ml in 0.25mol/lTris ,pH 8.0)和0.8mL0.25mol/l 的EDTA溶液,混匀后于37℃处理1h(2h或过夜),再加入200uL 10%SDS溶液,混匀后放置于55℃处理5min;⑤加入等体积的酚-氯仿(1:1),盖好,上下颠倒混匀3min,12000rpm,离心20min;⑥转移上相至新的离心管中,重复上个步骤直到无白色变性蛋白层出现;⑦取上清,加入3uL 10mg/ml的去DNase的RNase溶液,37℃水浴1h;(此步骤可省略)⑧重复步骤5、6⑨在上相中加入1/10体积的3mol/l NaAc(pH 5.2)和2倍体积的冷无水乙醇,-20℃放置30min;⑩ 4℃,12000rpm离心10min,弃上清,沉淀用70%乙醇清洗两次,干燥后溶于100ul的TE中,-20℃保存备用。
16SrDNA片段的PCR扩增1. PCR扩增16S rDNA序列以细菌基因组总DNA为模板,.扩增所用引物为:上游引物:5’-GAG AGT TTG ATC CTG GCT CAG-3’(27F)下游引物:5’-GGYTACCTTGTTACGACTT-3’(1492R)2.PCR体系(50μL体系)反应组分加入量模板(DNA) 1μLMgCl2(25mmol/L) 4μL10×缓冲液(Mg2+free) 5μLdNTP(10mmol/L) 4μL上游引物(10μmol/L)1μL下游引物(10μmol/L)1μLTaq酶(5u/μL)0.5μLH2O 33.5μL注:加Taq酶时要放在冰上操作2.反应条件94℃预变性2-5min(选5min),94℃30s,50-60℃(选55℃) 30s,72℃1min/1-2kb,30个循环;72℃ 5-10min(选10min),4℃保温。

1.16S rDNA-V3高变区的扩增1) 细菌基因组总DNA16S rDNA-V3高变区的扩增,50 µL体系:10×缓冲液(不含Mg2 +) 5 µLdNTP(10 mmol L-1) 1 µL引物341F、517R(10 μmol L-1)各0.5 µLMg2 + (25 mmol L-1) 2.5 µL普通Taq DNA聚合酶 2.5 UBSA(牛血清白蛋白1 mg mL-1) 5 µLDNA模板100 ngddH2O 补至50 µL2) PCR 扩增条件为:94℃ 5 min94 ℃ 1 min65℃ 1 min 降1℃72℃ 1 min94℃ 1 min 10个循环65℃ 1 min 降1℃72℃1min94℃1min55℃1min 20个循环72℃1min72℃10 min2.变性梯度凝胶电泳(DGGE)PCR产物用基因突变检测仪分析,PAGE(聚丙烯酰胺)胶浓度为8%(w/v),变性梯度为40%~60% (100%的变性剂为100 mL溶液中含有42 g的尿素和40 mL的甲酰胺)。
a)配制40%变性溶液:试剂8%胶40%丙烯酰胺/二聚丙烯酰胺20 mL50×TAE 缓冲液 2 mL甲酰胺(去离子)16 mL尿素16.8 gddH2O 补至100 mLb)配制60%变性溶液:试剂8%胶40%丙烯酰胺/二聚丙烯酰胺20 mL50×TAE 缓冲液 2 mL甲酰胺(去离子)24 mL尿素25.2 gddH2O 补至100 mLc)按说明书组装梯度制胶器;d)在电泳槽中装入7 L 1×TAE 电泳缓冲液;e)取40%和60%变性溶液各15 mL,分别加入过硫酸铵(10%)150 µL,TEMED15 µL,混匀,灌胶;f)标记为低浓度(L)和高浓度(H)的注射器分别吸入全部40%和60%变性胶溶液,通过一系列连接装置,按说明书灌至自上而下浓度由低到高的连续梯度凝胶;g)小心插入梳子,让凝胶聚合大约一个小时,并把电泳控制装置打开,预热电泳缓冲液到60℃;h)PCR产物用PCR产物纯化试剂盒进行纯化,用注射针进样;i)电泳条件:60℃,40 V,20 min 预电泳;150 v,约7 h;j)电泳完毕后,先拨开一块玻璃板,然后将胶放入盘中。

1目的建立和规范16SrDNA扩增操作程序。
2责任16SrDNA扩增实验人员。
3适用范围适用于单菌和基因组16SrDNA扩增操作。
4术语和定义无5操作步骤4.1试剂:10× PCR Buffer;dNTPs;sgR;sgF;r-Taq酶;PCR模板:待测混合菌;ddH₂O。
4.2菌悬液制备:超净工作台灭菌后点燃酒精灯,取出灭过菌的1.5mL离心管,做与菌号相应的编号,吸取30μL ddH₂O,在火焰附近用灭过菌的牙签或枪头挑取单个菌并于水中吸打混匀,使成有一定浊度的菌悬液。
4.3菌悬液的预处理:将菌悬液置-20℃冰箱中15min,取出,再将菌悬液置65℃水浴锅放10min;反复冻融三次后,在掌上离心机上离心10s。
4.4基因组样品的预处理:将提取好的基因组用灭过菌的ddH₂O稀释10倍,混匀离心,取其稀释液进行PCR扩增。
4.5配置样品PCR反应溶液4.5.1PCR反应体系组分1次反应所需用量(μL)n次反应所需用量(μL)ddH₂O 18.2 18.2×(n+1)10× PCR Buffer 2.5 2.5×(n+1)dNTP 2.0 2.0×(n+1)sgR 0.5 0.5×(n+1)sgF 0.5 0.5×(n+1)r-Taq酶0.3 0.3×(n+1)模板 1.0总体积25.04.5.2在超净工作台中,取1.5mL或2 mL离心管放置在冰盒上,根据PCR反应体系各溶液量,计算所需各溶液用量,按ddH2O、10× PCR Buffer、dNTP、sgR、sgF、r-Taq 酶的顺序加入离心管中(不加模板),配制完成后,振荡3秒或颠倒5次混匀,离心10秒。
4.5.3分装:PCR反应混合液(未加模板)配置后,用移液器吸取24μL分装至200μL的离心管中。
4.5.4加模板:将制备好的菌悬液(基因组稀释液)用移液器吸取1μL加入上述分装好的混合液中;其中一支分装后的PCR混合液加入1μL的纯化水做阴性对照。

16s rdna测序原理16s rDNA测序是一种用于研究微生物群落结构和功能的重要技术,它可以帮助科研人员了解微生物的多样性和相互关系,对环境微生物的研究具有重要意义。
本文将介绍16s rDNA测序的原理及其在微生物学研究中的应用。
16s rDNA是细菌和古细菌的小亚基RNA基因的一部分,它在所有细菌和古细菌中都存在,并且在细菌的进化过程中具有高度的保守性。
因此,通过对16s rDNA序列进行测序和比对,可以帮助科研人员了解不同微生物的分类和演化关系。
在进行16s rDNA测序时,首先需要从样品中提取微生物的DNA,然后通过PCR扩增得到16s rDNA的片段。
接下来,对PCR产物进行纯化和测序准备,最常用的方法是通过测序仪进行Sanger测序。
随着高通量测序技术的发展,现在也可以使用Illumina、454或Ion Torrent等平台进行高通量测序,大大提高了测序的效率和速度。
得到16s rDNA序列后,接下来的工作就是对序列进行比对和分析。
科研人员可以利用公开数据库中的16s rDNA序列作为参考,通过比对来确定待测序列的分类地位和系统发育关系。
此外,还可以利用一些生物信息学工具对序列进行多样性分析、物种丰度分析等,从而了解微生物群落的结构和功能。
在微生物学研究中,16s rDNA测序被广泛应用于环境微生物群落的研究。
通过对土壤、水体、空气等不同环境中微生物的16s rDNA进行测序和分析,可以揭示微生物的多样性、分布规律以及其对环境的影响。
此外,16s rDNA测序还可以用于研究人体内的微生物群落,例如肠道菌群的研究,有助于了解微生物与宿主健康的关系。
总之,16s rDNA测序是一种重要的技术手段,它为科研人员提供了解微生物多样性、分类和系统发育关系的重要途径,对微生物学、生态学和生物医学研究具有重要意义。
随着测序技术的不断发展和完善,相信16s rDNA测序在微生物学研究中的应用将会更加广泛和深入。

16SrDNA鉴定菌株的标准操作规程1.适用范围本标准规定了通过特定引物对细菌的16SrDNA片段进行PCR扩增,然后对扩增片段进行序列分析比对,快速获得细菌种属信息的操作规程。
本标准适用于未知细菌的快速种属分析,以及为细菌的生化鉴定提供指导信息。
2.方法和原理16SrDNA鉴定是指用利用细菌16SrDNA序列测序的方法对细菌进行种属鉴定。
包括细菌基因组DNA提取、16SrDNA特异引物PCR扩增、扩增产物纯化、DNA测序、序列比对等步骤。
是一种快速获得细菌种属信息的方法。
细菌rRNA(核糖体RNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。
16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列。
16S rDNA由于大小适中,约1.5Kb左右,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,故被细菌学家和分类学家接受。
在16S rRNA 分子中,可变区序列因细菌不同而异,恒定区序列基本保守,所以可利用恒定区序列设计引物,将16S rDNA片段扩增出来,利用可变区序列的差异来对不同菌属、菌种的细菌进行分类鉴定。
16SrDNA序列的前500bp序列变化较大,包含有丰富的细菌种属的特异性信息,所以对于绝大多数菌株来说,只需要第一对引物测前500bp序列即可鉴别出细菌的菌属。
针对科学论文发表或是前500bp无法鉴别的情况,需要进行16SrDNA的全序列扩增和测序,得到较为全面的16SrDNA的序列信息。
由于测序仪一次反应最多只能测出700bp的有效序列,为了结果的可靠性,通常将16SrDNA全长序列分成3部分进行测序。
3.设备和材料3.1器材移液器(1000μL、200μL 、100μL、10μL);涡旋振荡器;Eppendorf MixMate;离心机;水浴锅;电泳仪;制冰机;低温冰箱;PCR仪:Veriti 96 Well Thermal Cycler;凝胶成像仪:VersaDoc MP 4000;基因分析仪:AB3500、AB31303.2试剂DNA快速提取试剂:PrepMan Ultra;琼脂糖;PCR试剂:Taq酶,10×Taq Buffer(Mg2+),dNTPs,O等; ExoSAP-IT;测序试剂:BigDye Terminator,5×Sequencing Buffer;BigDye XTerminator ddH2Purification Kit;3.3耗材移液器吸头:1000μL、200μL、10μL;离心管:1.5mL、200μL;Micro Amp TM Optical 96-Well ReactionPlate;Micro Amp TM Optical Adhesive Film;3.4引物16SrDNA 名称序列扩增长度第1部分正向引物27F5'-AGA GTT TGA TCC TGG CTC AG-3'500 bp左右反向引物519R5'- GWA TTA CCG CGG CKG CTG -3'第2部分正向引物357F5'- CTC CTA CGG GAG GCA GCA G-3'750bp左右反向引物1115R5'-AGG GTT GCG CTC GTT GC-3'第3部分正向引物926F5'- AAA CTY AAA KGA ATT GAC GG-3'560 bp左右反向引物1492R5'-TAC GGC TAC CTT GTT ACG ACT T-3'其中M=C:A, Y=C:T. K=G:T, R=A:G, S=G:C. W=A:T; all 1:1 4.操作流程5.实验方法5.1 核酸提取:挑取单菌落,然后置于装有100μLPrepMan Ultra 的离心管中,涡旋震荡混匀30s 左右,然后100℃水浴10min 后,以离心机最大转速离心3min ,取10μL 上清液与490μL ddH 2O (即稀释50倍),混匀作为下步PCR 的模板DNA 。

16s收样标准
16S rDNA测序是指选择16S rDNA某个或某几个变异区域,选择通用引物对环境样本(肠道、土壤、水体等)微生物进行PCR扩增,然后对PCR产物进行高通量测序,并将得到的测序数据与已有的16S rDNA数据库进行比对分析,从而对环境群落多样性进行研究,核心是物种分析,包括微生物的种类,不同种类间的相对丰度,不同分组间的物种差异以及系统进化等。
16S rDNA的标准样品的制备通常需要经过以下几个步骤:
1. 样品收集:根据研究目的,选择适当的样品类型和收集方法。
例如,土壤、水体、粪便等环境样品中可能含有丰富的微生物多样性。
2. 样品处理:将样品进行适当的预处理,以提取出其中的微生物DNA。
这
可能包括破碎细胞、去除杂质、纯化DNA等步骤。
3. 16S rDNA扩增:使用特定的引物对样品中的16S rDNA进行PCR扩增,以获得足够的DNA片段供后续分析。
4. 测序:将扩增后的16S rDNA片段进行高通量测序,获得每个样品的测
序数据。
5. 数据分析:对测序数据进行处理和分析,包括去噪、质量控制、物种分类和相对丰度分析等。
在制备标准样品时,需要注意以下几点:
1. 选择适当的样品类型和收集方法,以保证样品的代表性和多样性。
2. 在处理样品时,要充分去除杂质和抑制物,以避免对后续PCR扩增和测序的干扰。
3. 选择适当的引物和PCR条件,以保证扩增的特异性和效率。
4. 对测序数据进行质量控制和预处理,以获得可靠的分析结果。
5. 对分析结果进行适当的解读和解释,以了解样品中的微生物群落结构和多样性。

一、实验原理随着分子生物学的迅速发展,细菌的分类鉴定从传统的表型、生理生化分类进入到各种基因型分类水平,如(G+C)mol%、DNA杂交、rDNA指纹图、质粒图谱和16S rDNA序列分析等。
细菌中包括有三种核糖体RNA,分别为5S rRNA、16S rRNA、23S rRNA,rRNA基因由保守区和可变区组成。
16S rRNA对应于基因组DNA上的一段基因序列称为16S rDNA。
5S rRNA虽易分析,但核苷酸太少,仅几十bp,没有足够的遗传信息用于分类研究;23S rRNA含有的核苷酸数几乎是16S rRNA的两倍,分子量太大,分析较困难。
而16S rRNA相对分子量在2kb左右,较为适合PCR 扩增,又具有保守性和存在的普遍性等特点,序列变化与进化距离相适应,序列分析的重现性极高,因此,现在一般普遍采用16S rRNA作为序列分析对象对微生物进行测序分析。
16SrRNA的编码基因是16SrDNA,但是要直接将16SrRNA提取出来很困难,因为易被广泛存在的RNase降解,因而利用16S rDNA鉴定细菌,其技术路线如下:PCR实验原理即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。
是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA。
二、主要器具及试剂PCR、电泳系统、DNA提取体系、Taq Polymerase、DNA Marker,溶菌酶、dNTP和E.coli JM109感受态细胞、pMD18-T Vector、琼脂糖、SDS裂解缓冲液、50×TAE电泳缓冲液贮存液、1×TE(pH 8.0)三、操作方法1. 细菌基因组总DNA的提取接种纯化的菌株于LB液体培养基中,180 r/min,37 ℃培养过夜,按以下的方法提取细菌基因组总DNA。

16SrDNA测序分析流程
宏基因组测序是指对微生物群体进行高通量测序,分析特定环境中微生物群体基因组成及功能、微生物群体的多样性与丰度,进而分析微生物与环境、微生物与宿主之间的关系,发现具有特定功能的基因。
宏基因组测序无需分离纯培养微生物,较大扩展了微生物资源的利用,为环境微生物群落的研究提供了有效工具。
宏基因组深度测序可以揭示或估计环境中真实的物种多样性和遗传多样性,挖掘具有应用价值的基因资源,应用于开发新的微生物活性物质。
宏基因组研究分两个方向:扩增子测序和全基因组测序。
扩增子测序,涉及特定序列位点的PCR扩增,通常是16S/18S rDNA。
宏基因组的物种分类,一般用OUT(operational taxonomic unit),即可操作单元来表示。
通常原生生物使用16S rDNA来衡量,真核生物的OUT使用18S rDNA来衡量。
主要应用在以下几个方面:通过宏基因组测序的方法,将肠道微生物与疾病进行关联分析以揭示疾病与健康个体间的微生物差异;鉴定特定环境中的特定微生物发现耐受菌种及相关基因。
可以研究物种的分类,研究与特定环境相关的代谢通路,以及通过不同样品的比较研究微生物群落内部、微生物与环境、微生物与宿主之间的关系。
扩增子序列(16rDNA)分析流程:
结果示例:
图1 不同样本OUT(operat ional taxonomic units)丰度的热图展示
图2 实验组和对照组在属水平上生物种类分布的柱状图和饼状图
图3 系统发生树。

16s rdna测序原理16S rDNA测序是一种常用的微生物多样性分析方法,通过对16S rDNA基因的测序和分析,可以揭示微生物群落的组成和结构,对环境微生物的研究具有重要意义。
本文将介绍16S rDNA测序的原理及相关内容。
1. 16S rDNA基因简介。
16S rDNA是细菌和古细菌的小亚基核糖体RNA基因,其序列在细菌中高度保守,但又存在一定的变异性,这使得16S rDNA成为研究微生物系统发育和分类的理想分子标记。
在细菌和古细菌中,16S rDNA一般由9个高度保守的区域(称为conserved region)和10个变异区域(称为variable region)组成,其中变异区域的序列差异较大,可用于微生物的分类和鉴定。
2. 16S rDNA测序原理。
16S rDNA测序的原理是通过PCR扩增获得16S rDNA基因片段,然后对扩增产物进行测序,最后对测序结果进行分析和解读。
首先,从样品中提取微生物DNA,然后利用通用或特异引物对16S rDNA基因进行PCR扩增,得到所需的16S rDNA片段。
接下来,对PCR产物进行纯化和测序,通常采用Sanger测序法或高通量测序技术(如Illumina、454、Ion Torrent等)。
最后,利用生物信息学方法对测序结果进行分析,包括序列比对、物种注释、多样性分析等。
3. 16S rDNA测序分析。
在16S rDNA测序分析中,首先需要对测序结果进行质控和过滤,去除低质量序列和引物污染,然后进行序列比对和物种注释。
序列比对是将测序结果与16S rDNA数据库进行比对,找到最佳匹配的参考序列,从而确定微生物的分类和系统发育关系。
物种注释是根据比对结果,将未知序列注释为已知的微生物分类单元,如属、种等。
此外,还可以进行多样性分析,如Alpha多样性指数(反映微生物群落的丰富度和多样性)、Beta多样性分析(比较不同样品间的微生物群落差异)等。


16s rDNA序列是细菌和古细菌特有的一种特征序列,是通过测定16s rDNA基因所编码的16s rRNA的序列而得到的。
在分子生物学和微生物学领域,16s rDNA序列被广泛应用于微生物分类、微生物多样性研究和微生物系统进化研究中。
本文将从以下几个方面对16s rDNA 序列进行介绍和分析。
一、成因和结构16s rDNA序列是细菌和古细菌特有的一种特征序列,它是细菌和古细菌核糖体小亚基rRNA基因的一部分,通常有约1500个核苷酸碱基对,可由16s rRNA基因编码。
这一序列在细菌和古细菌的核糖体RNA中起着重要的作用,它能够稳定地与核糖体蛋白结合,形成核糖体的小亚基,并参与到细菌和古细菌的蛋白质合成过程中。
二、意义和应用1. 微生物分类16s rDNA序列在微生物分类中具有重要意义,通过对16s rDNA序列进行测序分析可以鉴定和分类细菌和古细菌的种属和亚属。
这是因为16s rDNA序列在不同种属和不同亚属的细菌和古细菌之间存在一定的变异,可以作为分子生物学特征用于分类鉴定。
2. 微生物多样性研究通过对环境样品中的16s rDNA序列进行测序分析,可以了解微生物裙落的组成和结构,揭示微生物在自然界的分布和多样性。
这对于研究微生物的生态学、环境适应性和生态功能具有重要意义。
3. 微生物系统进化研究利用16s rDNA序列进行系统进化研究,可以揭示细菌和古细菌的系统发育关系和演化过程,为了解细菌和古细菌的起源、多样性和进化提供重要的分子学证据。
三、研究方法1. PCR扩增通常情况下,从细菌或古细菌的DNA中提取16s rDNA序列,然后利用PCR技术进行扩增。
通过选择适当的引物和反应条件,可以特异性地扩增出16s rDNA序列,为后续的测序分析做准备。
2. 测序分析测序是获取16s rDNA序列信息的关键步骤,目前常用的测序方法包括Sanger测序和高通量测序。
通过测序分析,可以获得16s rDNA 序列的具体碱基序列信息,用于后续的分类鉴定和系统进化研究。


16s rdna测序原理16s rDNA测序是一种用于研究微生物群落结构和多样性的重要技术。
它通过对细菌和古菌16s rDNA基因的测序,可以揭示微生物的分类和系统发育关系,帮助科学家更好地了解微生物在自然界中的分布和功能。
下面,我们将详细介绍16s rDNA测序的原理及其在微生物学研究中的应用。
首先,我们需要了解16s rDNA是什么。
16s rDNA是细菌和古菌细胞中的一个重要基因,它编码了16s rRNA,是细菌和古菌的核糖体小亚基的组成部分。
由于16s rDNA在不同的细菌和古菌中存在一定的差异,因此可以作为分类和鉴定微生物的分子标记。
通过对16s rDNA序列的测定和比对,可以揭示微生物的遗传关系和系统发育地位。
在进行16s rDNA测序时,首先需要从样品中提取微生物的总DNA。
接下来,利用特异性引物对16s rDNA基因进行PCR扩增,得到16s rDNA的扩增产物。
然后,对扩增产物进行测序,获取16s rDNA的序列信息。
最后,利用生物信息学方法对测得的16s rDNA序列进行分析和比对,从而揭示微生物的分类和系统发育关系。
在微生物学研究中,16s rDNA测序被广泛应用于微生物群落结构和多样性的研究。
通过对不同环境样品中微生物16s rDNA序列的测定和分析,可以揭示微生物在不同环境中的分布和多样性,帮助科学家更好地了解微生物在自然界中的生态功能。
此外,16s rDNA测序还可以用于鉴定和分类未培养的微生物,为微生物资源的开发和利用提供重要的信息。
总之,16s rDNA测序是一种重要的技术手段,可以帮助科学家揭示微生物的分类和系统发育关系,了解微生物在自然界中的分布和功能。
随着测序技术的不断发展和进步,相信16s rDNA测序在微生物学研究中会发挥越来越重要的作用,为我们认识微生物世界提供更多的信息和启示。

16s rdna测序原理16S rDNA测序是一种基于16S rRNA基因的测序方法,该方法用于研究微生物的多样性和进化关系。
16S rRNA基因存在于所有细菌和古细菌的基因组中,它具有高度保守的序列区域和变异的序列区域,可以作为鉴定和分类微生物的分子标记。
16S rDNA测序的原理是通过PCR扩增目标DNA片段,然后将扩增的DNA片段纯化,接着进行测序。
一般来说,测序过程包括DNA提取、PCR扩增、测序反应、分析和比对等步骤。
首先,在样品中提取出微生物的总DNA,包括细胞外和细胞内的DNA。
提取方法根据样品的类型和要求的纯度进行选择,常用的是酚-氯仿法或商业DNA提取试剂盒。
然后,选择适当的引物对16S rDNA进行PCR扩增。
引物的选择需要考虑到引物的特异性和适用范围,常用的引物是通用的16S rDNA引物,例如27F和1492R。
PCR扩增可以通过热循环法进行,其中包括一系列的加热、退火和延伸步骤。
接下来,对PCR扩增得到的DNA片段进行纯化处理,去除PCR反应中的引物和杂质,以获得纯净的目标DNA片段。
纯化方法可以使用商业的DNA纯化试剂盒或凝胶纯化等。
然后,对纯化后的DNA片段进行测序反应,通常使用链终止法(Sanger测序)或高通量测序技术(如Illumina MiSeq或Ion Torrent)进行。
测序反应产生的测序片段将通过鉴定碱基的荧光信号或电信号来确定碱基的顺序。
最后,通过分析软件对测序结果进行分析和比对,将测序片段与已知的16S rDNA序列数据库进行比对,以识别样品中存在的微生物并进行分类和系统发育分析。
常用的分析软件包括QIIME、Mothur和MEGAN等。
16S rDNA测序方法已成为研究微生物多样性和进化关系的重要工具,其原理简单且经济高效,可以在较短的时间内获得大量的序列数据,为深入了解微生物的生态学、进化和功能研究提供了有效的手段。

16S rDNA鉴定细菌的方法细菌16S rDNA鉴定主要分为7个部分:1.提取细菌基因组DNA,2.设计/选择引物进行PCR扩增,电泳检测纯度与大小。
3.琼脂糖凝胶电泳分离4.胶回收目的片段5.目的片段测序。
6.BLAST比对获取相似片段。
7.构建系统进化树试剂:1、培养基:通常选择组分简单且细菌生长良好的培养基(培养基组分过于复杂会影响DNA 的提取效果,也可以在裂解细菌前用TE缓冲液对菌体进行洗涤。
)。
2、1M Tris-HCl (pH7.4, 7.6, 8.0)(1L):121.1g Tris,加浓盐酸约(70ml, 60ml, 42ml),高温高盐灭菌后,室温保存。
冷却到室温后调pH,每升高1℃,pH大约下降0.03个单位。
3、0.5M EDTA(pH8.0)(1L):186.1g Na2EDTA•2H2O,用NaOH调pH至8.0(约20g),高温高压灭菌,室温保存。
4、10×TE Buffer(pH7.4,7.6,8.0)(1L):组分:100 mM Tris-HCl,10 mM EDTA。
1M Tris-HCl (pH7.4,7.6,8.0)取100ml,0.5M EDTA(pH8.0)取20ml。
高温高压灭菌,室温保存。
1×TE Buffer用10×TE Buffer稀释10倍即可。
5、10%SDS(W/V):称10g,68℃加热溶解,用浓盐酸调pH至7.2。
室温保存。
用之前在65℃溶解。
配置时要戴口罩。
6、5M NaCl:称292.2gNaCl,高温高压灭菌,4℃保存。
7、CTAB/NaCl(10%CTAB,0.7M NaCl):溶解4.1g NaCl,加10g CTAB(十六烷基三甲基溴化铵),加热搅拌。
用之前在65℃溶解。
8、氯仿/异戊醇:按氯仿:异戊醇=24:1(V/V)的比例加入异戊醇。
9、酚/氯仿/异戊醇(25:24:1):按苯酚与氯仿/异戊醇=1:1的比例混合Tris-HCl平衡苯酚与氯仿/异戊醇。
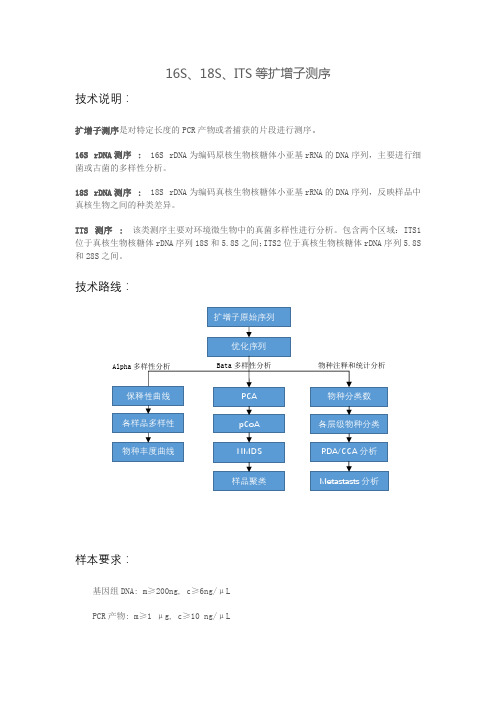
16S 、18S 、ITS 等扩增子测序技术说明:扩增子测序是对特定长度的PCR 产物或者捕获的片段进行测序。
16S rDNA 测序 : 16S rDNA 为编码原核生物核糖体小亚基rRNA 的DNA 序列,主要进行细菌或古菌的多样性分析。
18S rDNA 测序 : 18S rDNA 为编码真核生物核糖体小亚基rRNA 的DNA 序列,反映样品中真核生物之间的种类差异。
ITS 测序 : 该类测序主要对环境微生物中的真菌多样性进行分析。
包含两个区域:ITS1位于真核生物核糖体rDNA 序列18S 和5.8S 之间;ITS2位于真核生物核糖体rDNA 序列5.8S 和28S 之间。
技术路线:样本要求:基因组DNA: m ≥200ng, c ≥6ng/μLPCR 产物: m ≥1 μg, c ≥10 ng/μL扩增子原始序列优化序列PCA pCoA NMDS样品聚类 物种分类数 各层级物种分类RDA/CCA 分析 Metastasts 分析保释性曲线各样品多样性物种丰度曲线 Alpha 多样性分析 Bata 多样性分析 物种注释和统计分析服务周期:标准流程的服务周期约为 30-55 个工作日。
案例解析:案例一Defining the core Arabidopsis thaliana root microbiome. [ Nature,2012]1.背景介绍植物根部微生物生存部位可分为根内、根周边和土壤。
其中,根内和根周边微生物对植物生长、生产、碳源固定和植物修复等有重要作用。
植物根部虽然有免疫系统,依然允许一些互惠共生微生物的生存。
植物对根部微生物的生存的调控机制,现在还不是很清楚。
通过16S对微生物种类和数量的分析,来验证根内和根周边微生物的类型,主要取决于植物本身与其基因型,与土壤类型和植物生长阶段无关。
首先,文章验证了,根内、根周边和土壤这三个位子的菌群结构有很大差异。
根内部菌群结构相对简单,主要以放线菌和变形菌为主。

细菌16srdna序列细菌16S rDNA序列是一种重要的遗传物质,它在细菌分类学和进化研究中扮演着重要的角色。
本文将从细菌16S rDNA序列的意义、应用以及分析方法等方面进行阐述。
一、细菌16S rDNA序列的意义细菌16S rDNA序列是细菌基因组中的一个高度保守的区域,它在细菌中普遍存在,具有高度的保守性和多样性。
因此,通过分析细菌16S rDNA序列,可以揭示细菌的亲缘关系和分类地位。
细菌16S rDNA序列的分析可以帮助我们了解细菌的进化关系、物种多样性以及细菌在环境中的分布情况。
二、细菌16S rDNA序列的应用1. 细菌分类和鉴定:通过比对已知的细菌16S rDNA序列数据库,可以确定未知细菌的分类地位和鉴定其物种。
这对于细菌的分类学研究以及临床微生物学和环境微生物学等领域具有重要意义。
2. 细菌进化研究:通过比较不同细菌的16S rDNA序列,可以推断它们之间的进化关系和亲缘关系,进而了解细菌的进化历史和演化过程。
3. 环境微生物学研究:通过分析环境样品中的细菌16S rDNA序列,可以了解细菌在不同环境中的分布情况,揭示细菌在自然界中的功能和作用。
三、细菌16S rDNA序列分析方法1. PCR扩增:首先需要从细菌中提取出基因组DNA,然后使用引物对16S rDNA序列进行PCR扩增。
PCR扩增是将16S rDNA序列扩增成大量的DNA片段,为后续的测序提供足够的模板。
2. 测序:将PCR扩增产物进行测序,可以使用Sanger测序或高通量测序技术。
测序得到的数据就是细菌16S rDNA序列的碱基信息。
3. 数据分析:将测序得到的数据与已知的16S rDNA序列数据库进行比对,使用比对软件进行序列比对和物种鉴定。
常用的比对软件有BLAST和MEGA等。
4. 构建系统发育树:根据比对结果,可以使用系统发育学的方法,如最大似然法或邻接法,构建细菌的系统发育树,揭示细菌之间的亲缘关系和进化关系。

16SrDNA
1.16SrDNA
是编码原核生物16SrRNA的基因,长度约1500bp,存在于所有细菌、衣原体、支原体、立克次体、螺旋体和放线菌等原核生物的基因组中,由多个保守区(conservedregion)和与之相间的多个可变区(variableregion)组成。
保守区为所有细菌共有,细菌之间无显著差异;可变区在不同细菌之间存在一定程度的差异,具有细菌属或种特异性。
PCR扩增16SrDNA包含两层含义:在保守区设计PCR通用引物,理论上可将存在于待测标本中的各种细菌都扩增出来;若选择可变区设计PCR特异性引物,则可将标本中的细菌鉴定到属、种乃至菌株水平。
因此,
16SrDNA已成为细菌鉴别与分类研究中较理想的靶序列。
2. 引物设计及扩增假阳性问题
尽管细菌16SrDNA存在多个保守区,但没有针对哪个保守区序列设计的PCR引物适合于扩增所有细菌。
细菌、衣原体、螺旋体、立克次体等不同种类微生物之间16SrDNA保守区虽然存在共有序列(consensussequence),但各种类间仍存在若干碱基不同。
扩增细菌16SrDNA的“通用引物”也是相对的,通常是在共有序列的基础上设计一套PCR引物。
因此,针对被检微生物的种类不同或临床标本不同,应选择相对应的通用引物,才能达到特异扩增效果。
如果引物设计不当,还可出现与人基因组的交叉反应,导致PCR结果错误判读。
由于PCR本身的高敏感度,极微量污染也可能出现假阳性。
污染可发生在标本采集、核酸提取乃至PCR操作各环节。
因此,在PCR扩增及其测序的各环节都必须建立并遵循标准化操作规程。

实验报告解析—微生物多样性(扩增子16SrDNA测序)1.实验报告的主体框架内容a)封面信息b)项目实施信息c)元基因组DNA提取过程及结果d)PCR扩增过程及结果e)样品是否成功的判定标准a)封面信息•主要包含:客、服双方的主要联系人相关信息,样品对接人,实验操作组及实验主要负责人信息,报告生成时间、项目名称及唯一标识号等方面内容。
•主要目的:用于追溯实验流程过程,客、服双方在实验方面内容的沟通及对接。
b)项目实施信息•主要包含:样品的详细情况(客户方提供的每例样品的名称、服务方提供的每例样品在服务方内部查询及流转的唯一名称、样品类型等方面内容),以及每例样品在实验后是否有剩余的统计情况。
•主要目的:让客户方了解每一个样本的基本情况;与服务方内部的对应关系,便于查询;实验操作后,样品的剩余情况或其他需要备注的特殊情况。
c)元基因组DNA提取过程及结果•主要包含:提取过程的核心步骤概括;参考文献或试剂盒使用说明书;DNA质量检测的方法;结果展示与汇总(DNA完整性的检测电泳图,浓度及纯度的测定结果,获取核酸溶液的总体积等必要信息)。
•主要目的:使得客户方了解提取的环节和细节;实验过程的最终情况及质控情况。
d)PCR扩增过程及结果•主要包含:扩增16srDNA的哪些区域,引物名称及引物序列;每例样品的稀释情况汇总(DNA溶液的消耗、稀释终浓度等);PCR 扩增最终反应体积及扩增条件;扩增结果检测电泳图及结果汇总等。
•主要目的:了解扩增过程实验详情、质控情况;样品实验成功及失败的情况,便于调整下一步实验计划和安排。
e)样品是否成功的判定标准•主要包含:对结果等级判别进行标准化并给予确切描述。
•主要目的:解释和说明结果成功或失败的原因。
2. 实验结果内容的体现及判定•实验结果内容体现:在报告中,此部分往往是通过汇总表格进行体现。
表格体现比较清晰且有条理。
因此,报告中表格部分,往往是每例样品的具体信息,若关注样品的具体情况,需要留意报告中的每一份表格内容。