黄药的分析方法总结
- 格式:docx
- 大小:40.15 KB
- 文档页数:5


黄药黄药(xanthate) 硫化矿浮选常用的一种巯基扩捕收剂。
学名为烃基黄原酸盐,通式,R为C2~5烷基。
醇与苛性碱和二硫化碳作用,生成黄药其基本反应式为性质黄药为黄色晶体或粉末,不纯品常为黄绿色或橙色的胶泥状物,有刺激性臭味,中等毒性,因此,生产黄药时应注意保护人体和防止环境污染。
短碳链黄药易溶于水,易燃,稳定性差,合成黄药含水分多,保存期为半年。
放置时间过长则结块变质,干燥黄药则比较稳定,能较长时间存放。
黄药在水中水解成黄原酸,溶液呈碱性:在酸性介质中黄原酸分解成醇和二硫化碳:黄药与重金属离子作用生成难溶性盐:式中Me2+为……等。
黄药被氧化则生成双黄药:合成方法黄药早在1782年即已被合成,用作分析试剂,直至1925年才用于浮选作捕收剂。
合成工艺有多种,如直接合成法、水溶液法、稀释剂法、部分稀释剂法、过量醇法、蒸汽法、碱金属醇淦法等。
中国采用直接合成法生产,利用强烈搅拌的捏和机及在冷冻的条件下,将理论比例量的醇与氢氧化钠粉末互相作用,再缓慢加入二硫化碳,进行黄原酸化反应,得合成黄药,经干燥得干燥黄药;也可以采用“反加料法”,即先将醇与二硫化碳混合,再慢慢有控制地加入氢氧化钠粉末制成黄药。
应用黄药用途甚广,迄今已有近70年的使用历史,在浮选工业中黄药用作硫化矿捕收剂,橡胶工业中用作硫化促进剂,分析化学中用乙基黄原酸钠作铜镍等金属离子的沉淀剂及比色剂,冶金工业中用黄药从溶液中沉淀钻镍,纤维素黄药用于制造人造纤维。
黄药适用于浮选铜、铅、锌等金属硫化矿时用作捕收剂,对某些氧化矿,如氧化铜矿、氧化铅锌矿,用硫化钠硫化后也可以黄药作捕收剂进行浮选。
浮选用的黄药有钾黄药和钠黄药两大类,在浮选中起捕收作用的是黄原酸根,与钾、钠离子关系不大,因此烃基相同的钾黄药或钠黄药有相同的选矿效能。
钠黄药在空气中较易吸湿受潮,但较便宜,中国均使用钠黄药。
黄药因其分子中的烃基不同,而有不同品种,常用的有乙基、正丙基、异丙基、正丁基、异丁基和戊基等黄药,它们共同的特点均为黄色晶体或粉状固体,亦可压成短条状或粒状出售,含黄原酸钠一般在77%以上,含游离碱0.5%以下,易溶于水。

浮选溶液中黄药及其分解产物的分析现状摘要:本文综述了国内外对选矿水溶液中黄原酸盐的分析测定方法。
阐述了紫外分光光度法、化学滴定法、气相色谱法、高效液相色谱法、毛细管电泳法的基本原理,优缺点及测定效果。
并指出黄原酸盐测试技术将向多种药剂同时测定的方向发展,对选矿厂药剂合理利用和分配具有重要的意义。
关键词:综述;黄原酸盐;测试技术;前言自1925年Keller首次在浮选过程中使用黄药作为捕收剂以来[1],关于黄药在浮选溶液中的变化规律,赋存状态的研究就倍受关注,因为这关系到黄药在浮选过程中的合理用量[2],自动化检测和控制,以及在选矿废水处理过程中具有重要的意义。
但由于矿浆中成分复杂,溶液中干扰因素多,黄药在浮选过程中分解产物繁多,仪器设备的限制,加上实验操作的精确性,使得分析过程难上加难,分析结果不尽人意。
近年来,随着现代科技的不断创新,技术的不断改进,对黄药及其衍生物测定的仪器方法更加精确和成熟。
本文总结了目前国内外对黄药及其衍生物在溶液中常用的测试手段,并对各方法进行了比较,对捕收剂等微量药剂在溶液中的测定具有重要的意义。
一紫外可见分光光度法紫外可见分光光度法在浮选研究中主要用于测定溶液中的低浓度浮选药剂,研究药剂与矿物作用产物的组成,某些调整剂在浮选过程中的作用,以及药剂吸附动力学等。
李文艳等[3]利用紫外可见分光光度计测定生产废水中乙基黄原酸钾的含量。
先过滤出废水中的不溶性物质后,以待测废水为背景样进行校正,直接测定吸光度,有效的消除了干扰,该方法检出限为0.01mg /L,方法的线性范围为0.04—18mg /L,水样测定的相对标准偏差为1.63%。
贺心然等[4]采用紫外分光光度(UV)法测定待测水样中丁基黄原酸浓度,用待测水样作为背景校正,并通过对不溶性物质,可溶性物质如硝酸盐,亚硫酸盐,以及金属离子的干扰实验,使得实际水样的测定相对标准偏差小于5.76%,检出限为0.006 mg/ L、测定上限为12.00 mg/ L,利用不同方法对样品进行分析测试,无明显差异。

水中超痕量黄药的顶部空间气相色谱法
水中超痕量黄药的顶部空间气相色谱法是一种用于测定非溶解性成分在水中存在量的重要方法。
水中超痕量黄药的顶部空间气相色谱法主要用来分析微量黄药在水体中的存在量,它可以精确测定微量黄药的含量,为环境保护工作提供有效的参考资料。
该法分析特定黄药的步骤:步骤一:对水样准备。
从水样中取一定量的水,加上一定的酯类溶剂来提取黄药;步骤二:进行油脂类黄淀粉、尿液和HCl/NaOH 处理。
这些处理将消除水样中其他组成成分带来的干扰;步骤三:将提取液进行0.45μm滤膜处理,再到1~5 ml的GC柱上进行分析;步骤四:以恒温温度(200-250℃)、恒流量(2.0 mL/min)和恒压(20 psi)作为控制,进行检测,用检测结果进行定量分析。
水中超痕量黄药的顶部空间气相色谱法特别适用于测定微量非溶解性物质,如药物、农药、抗生素等,具有高灵敏度、低污染,操作简便等优点。
它是一种非常有效的水质监测方法,所得结果能够反映出水体中微量黄药成分的种类及含量,从而为把控水质质量和污染及时排查提供参考。

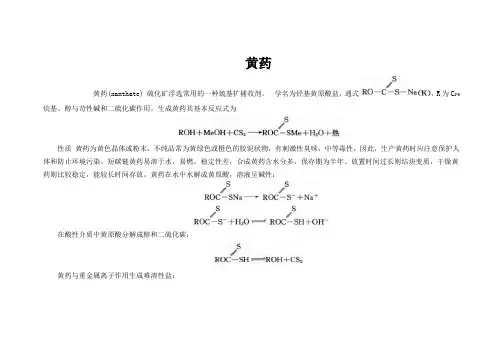
黄药xanthatehuangyao 黄药(xanthate)硫化矿浮选常用的一种筑基S / 捕收剂。
学名为烃基黄原酸盐,通式RO一C一S一Na (K),R为CZ_5烷基。
醇与苛性碱和二硫化碳作用,生成黄药其基本反应式为S / ROH+MeOH+CS:一ROC一SMe+HZO+热性质黄药为黄色晶体或粉末,不纯品常为黄绿色或橙色的胶泥状物,有刺激性臭味,中等毒性,因此,生产黄药时应注意保护人体和防止环境污染。
短碳链黄药易溶于水,易燃,稳定性差,合成黄药含水分多,保存期为半年。
放置时间过长则结块变质,干燥黄药则比较稳定,能较长时间存放。
黄药在水中水解成黄原酸,溶液呈碱性: SS Z/ ROC一SNa一ROC一S一十Na个SS // RO C一S一十HZO二二=乏ROC一SH+OH- 在酸性介质中黄原酸分解成醇和二硫化碳: S / ROC一SH节二二二亡ROH+CSZ 黄药与重金属离子作用生成难溶性盐: SS // ZROC一S Na+Mez十一(ROC一S)ZMe十+ZNa十式中MeZ+为CuZ+、PbZ+、ZnZ+、FeZ+……等。
黄药被氧化则生成双黄药: S / 4ROC一SNa+02十ZHZO一SS 尹尹ZROC一S一S一C一OR+4NaOH 合成方法黄药早在1782年即已被合成,用作分析试剂,直至1925年才用于浮选作捕收剂。
合成工艺有多种,如直接合成法、水溶液法、稀释剂法、部分稀释剂法、过量醇法、蒸汽法、碱金属醇淦法等。
中国采用直接合成法生产,利用强烈搅拌的捏和机及在冷冻的条件下,将理论比例量的醇与氢氧化钠粉末互相作用,再缓慢加入二硫化碳,进行黄原酸化反应,得合成黄药,经干燥得干燥黄药;也可以采用“反加料法”,即先将醇与二硫化碳混合,再慢慢有控制地加入氢氧化钠粉末制成黄药。
应用黄药用j兔甚广,迄今已有近70年的使用历史,在浮选工业中黄药用作硫化矿捕收剂,橡胶工业中用作硫化促进剂,分析化学中用乙基黄原酸钠作铜镍等金属离子的沉淀剂及比色剂,冶金工业中用黄药从溶液中沉淀钻镍,纤维素黄药用于制造人造纤维。


药物检验分析经验分享1、检验吲哒帕胺片的含量测定时,采用超声波超声可使片剂更易分散,主成分溶解完全,没有浪费,与研磨转移方法比较,对含量均匀度测定没有影响,且简单方便且更合理。
2、乙醇溶解主成分后,不能溶解辅料,需要过滤。
采用离心方法使辅料沉淀,取上清夜。
(注意,有很多离心管不具塞子,可用柔软的塑料薄膜袋扎橡皮筋做塞子。
没有塞子离心,偏差可达5%),与薄膜过滤法比较,对测定结果没有影响。
而且,如果过滤法操作不够快速,乙醇挥发,易影响测定结果。
3、在做中药材的浸出物的检测时,一定要按标准控制好溶剂的浓度(如乙醇等),否则检验结果会差异很大。
4、当液相鉴别中供试品与对照品出峰时间不一致,无法判断是否合格时,可用对照品与供试品各半配成混合溶液后进样,若峰宽未变宽,未出现驼峰,即可判断为合格。
5、做原料残留物检测的时候,如果主药对杂质有干扰,现有方法无法检出,需要自己建方法的话,要优先考虑利用理化性质将杂质分离出来再进行测定。
往往有意想不到的效果。
6、在药材薄层色谱鉴别时应该考虑一下展开剂的温度与配制顺序,有时会影响色谱的结果。
7、做药品有机溶剂残留量检查时,可以不拘泥于规定的色谱柱, 通常的DB-624可以满足要求,取样量也可以灵活调整,标准溶液浓度根据限度做相应调整就可以了。
8、薄层色谱鉴别时饱和时间一定要够。
9、在采用HPLC法测物质含量时,如若流动相中用了缓冲盐,一定要注意其pH值,放置过程中可能会产生变化,而某些药物对这种变化很敏感。
10、用HPLC法测物质含量时,温度的控制极其重要,最好用有柱温箱的,如果没有就要装空调保持恒温后再测定,否则会出现基线飘移,结果不准确。
11、有些品种温度的影响非常大,我遇到一个品种.对照品溶液不能放在冰箱中,放在液相室内还开着空调,第二天才能做,否则第二天的对照品到中午也不能用。
12、在使用微量移液枪时,要注意"重压轻打",加液会更家准确。

化学反应法鉴别大黄的原理大黄是一种常用的中药材,具有通便、清热、泻火、解毒等功效。
在中药饮片生产和质量控制过程中,鉴别大黄的原理常使用化学反应法。
化学反应法鉴别大黄的原理基于大黄中所含有的特定化学成分对特定试剂发生反应,从而通过观察反应产物的特征来判断是否为大黄。
以下为常见的几种化学反应法鉴别大黄的原理:1. 蒽醌试验法:大黄中含有丰富的大黄素,蒽醌试验法是利用大黄素与硫酸、硝酸等试剂发生氧化反应,产生特征性的颜色变化进行鉴别。
将大黄粉末与硫酸混合,生成红色、橙色或紫色的反应产物,可以判断样品中是否含有大黄。
2. 雷公藕皮试验法:大黄中的蒽醌类物质与亚甲基蓝等试剂发生氧化反应,产生特征性的绿色颜色变化。
将大黄粉末与亚甲基蓝溶液混合,观察是否出现绿色反应产物,即可鉴别大黄的存在。
3. 高锰酸钾试验法:大黄中的芳香族化合物与高锰酸钾溶液反应生成深紫色沉淀。
将大黄粉末与高锰酸钾溶液反应,观察是否出现深紫色沉淀来判断样品中是否含有大黄。
4. 焦亚硝酸试验法:利用大黄中的酚类物质与焦亚硝酸钠反应生成紫红色化合物。
将大黄粉末与焦亚硝酸钠溶液反应,观察是否出现紫红色化合物,即可鉴别大黄的存在。
以上的化学反应法鉴别大黄的原理基于大黄中特定物质的存在和其与试剂之间的反应所产生的特征性的颜色、沉淀等变化。
这些反应具有较高的选择性和特异性,能够准确判断样品中是否含有大黄。
需要注意的是,化学反应法鉴别大黄虽然简便易行且成本较低,但对于样品的前处理和试剂的选择要求较高,也需要注意反应条件的控制,以确保鉴别结果的准确性。
此外,化学反应法仅能判断样品中是否含有大黄,对于大黄的质量和纯度鉴定需要综合考虑其他方法,如色谱法、质谱法等。
总之,化学反应法通过观察大黄样品与特定试剂反应所产生的特征性变化,来鉴别大黄的存在。
这些方法简单易行且成本较低,为中药饮片生产和质量控制提供了一种有效的手段。

大黄药材含量测定方法一、背景介绍大黄是一种中药原料,广泛应用于中医临床和民间传统医药。
大黄常用于清热泻火、通便利肠等疾病的治疗。
大黄中的主要有效成分是大黄素,其含量水平是影响其药效的一个重要指标。
准确地测定大黄中大黄素含量非常重要。
下面我们将介绍10种关于大黄药材含量测定方法的详细描述。
二、主要测定方法1. 高效液相色谱法(HPLC)HPLC是一种常用的大黄素含量测定方法。
该方法具有准确、快速、灵敏度高、重复性好等优点。
HPLC方法要求使用高品质的设备,例如高速离心机,从而获得准确的分离和定量数据。
2. 紫外分光光度法(UV)UV法是一种常用的大黄素含量测定方法。
该法无须特殊试剂,分析成本低廉。
但该方法受杂质干扰大,精密度差,显色反应不稳定等因素的影响,因此使用前应先去除大黄中的杂质。
3. 燃烧法燃烧法是大黄素中含量测定的一种传统方法。
该方法常用于水分及灰分含量的测定,并且操作简单,使用方便。
该方法的主要限制是无法区分大黄素与其他化合物,因此需要与其他方法结合使用。
4. 比色法比色法是大黄素含量测定的另一种常用方法。
使用比色法可以直接测量大黄素的吸光度,从而得出含量。
该方法的限制是对其他化合物不敏感,因此只能测定大黄中的大黄素含量。
5. 红外光谱法红外光谱法是一种非常灵敏和准确的大黄素含量测定方法。
该方法可以检测大黄素的特征峰,从而获得准确的含量数据。
使用该方法需要使用高品质的光谱仪,同时精确控制样品的温度和物质状态,以避免实验中的误差。
6. 气相色谱法气相色谱法是一种常用的大黄素含量测定方法。
该方法可以通过大黄素在气相中的相对分离和挥发率来推断其含量。
该方法需要使用高品质的气相色谱仪,从而获得准确的分离和定量数据。
7. HPLC-质谱法HPLC-质谱法是一种非常灵敏和准确的大黄素含量测定方法。
该方法能够对大黄素进行鉴定和测定,避免假阳性和假阴性结果,提高测定的准确性。
使用该方法需要使用高品质的质谱仪和高效液相色谱仪。

书山有路勤为径,学海无涯苦作舟黄药的一般性质黄药是淡黄色的粉状物,因而得名。
易溶于水,应用时视选厂的具体情况,可配成1—10%的水溶液使用。
用量一般在50—150 克/吨。
黄药有下列一些性质值得注意。
1、离解、水解和分解黄药是黄原酸钠盐或钾盐,在水中溶解度较大。
并且发生电离;黄原酸盐是弱酸盐,在水中容易水解生成部分黄原酸,黄原酸在酸性介质中容易分解。
黄药在水中离解、水解和分解的反应,可用下面的反应式表示:ROCSSNa=ROCSS-+Na+黄药电离ROCSS-+H2O←→ROCSSH+OH-黄原酸根水解ROCSSH→CS2+ROH黄原酸分解ROCSSH←→ROCSS-+H+黄原酸电离某些黄原酸电离常数的负对数(PKa)和分解常数列于表1 中。
从表中1 看出,黄原酸是比较弱的酸,其PKa 值在2~3 间,故黄原酸根有水解作用。
从分解常数来看,一般说来,分子量愈大的,在水溶液中的分解常数愈小。
换句话说,分子量愈大的黄原酸,其水溶液愈稳定。
一般认为,PH﹤7 时,黄原酸根会水解变成黄原酸,生成的黄原酸进一步分解为醇和二硫化碳。
2、氧化在PH=7~12 时,黄药在水溶液中被氧化为双黄药。
几种黄原酸的pKa 值和分解常数表1 黄原酸名称黄原酸结构式pKa 值分解常数溶剂甲黄原酸乙黄原酸异丙基黄原酸丁基黄原酸戊基黄原酸异戊基黄原酸甲黄原酸乙黄原酸异丙基黄原酸丁基黄原酸戊基黄原酸异戊基黄原酸壬基黄原酸2.292.742.163.032.962.852.573.035.182.162.402.952.170.0240.0330.0270.0180.011 0.0120.9030.6890.3960.8630.6990.6160.643 水水水水水水二甲基甲酰胺双黄药也是硫化矿捕收剂,其选择性比黄药好。
[next] 3、在PH=7~12 时,黄药在水溶液中直接分解,一般认为PH 值小于7 时,黄药分解为醇和二硫化碳,当PH=7~12 时,被溶于水中的氧氧化为双黄药;有人认为,这是不全面的,除。
大黄药材含量测定方法(一)大黄药材含量测定大黄是一种常见的中药材,广泛应用于临床和家庭中。
为了保证大黄质量的稳定性和安全性,需要对其含量进行测定。
下面介绍几种测定大黄药材含量的方法。
1. 高效液相色谱法高效液相色谱法是一种常用的测定药材含量的方法。
该方法使用高效液相色谱仪,在药材中检测大黄的主要有效成分——大黄素的浓度。
该方法简单、准确,结果可靠。
2. 紫外-可见光谱法紫外-可见光谱法也是一种常用测定药材含量的方法。
该方法利用紫外-可见光吸收的原理,检测药材中大黄素的浓度。
该方法精确度高,同时操作简单。
3. 酸碱滴定法酸碱滴定法是一种传统的药材含量测定方法。
该方法利用化学反应原理,测定药材中的总黄酮含量。
虽然该方法操作简单,但其结果稳定性不如前两种方法。
4. 液相色谱法液相色谱法也是一种常用于药材含量测定的方法。
该方法利用高效液相色谱仪,对药材中大黄素含量进行测定。
该方法精确度高,结果可靠。
5. 薄层色谱法薄层色谱法是一种较为简单的药材含量测定方法。
该方法使用薄层色谱仪,对药材中大黄素含量进行测定。
该方法操作简单,同时费用较少。
总结以上几种方法均可用于测定药材中大黄素的含量,其中高效液相色谱法和紫外-可见光谱法具有较高的精确度,结果可靠,但费用较高。
而酸碱滴定法、液相色谱法和薄层色谱法则操作相对简单,费用较少。
需要根据实际需要选择适合的方法进行测定。
注意事项在进行大黄药材含量测定时,需要注意以下事项:1.样品准备:药材样品应当储存干燥、防潮、防虫害,并保持完整无损。
在测定前需要先对样品粉碎、筛选,并按照规定方法提取大黄素。
2.标准物质:在测定大黄素的含量时需要使用标准品来进行校准,确保测定结果的准确度和可靠性。
3.仪器操作:大黄药材含量测定需要配备相应的仪器设备,操作人员需要经过专门培训,熟悉仪器的使用方法及操作规程,以保证测定结果的准确性。
4.数据处理:在对测定结果进行处理时,需要正确使用统计学方法和软件来进行数据处理和分析,确保结果的准确性和可靠性。
黄精及其炮制品的薄层鉴别研究一、概述黄精是一种常用中药材,其性质温热,味苦。
在中医中,黄精被认为有养血安神、益气健脾的功效。
黄精广泛用于中医药、保健品等领域,由于错误的采收、储存和加工等环节,有些黄精可能被掺杂或夹杂着杂质,也可能被人为掺假,这就需要对黄精进行鉴别。
薄层分析法是一种简单、可靠、快速的分析技术,可以用来鉴别黄精及其炮制品。
二、黄精及其炮制品的薄层鉴别的方法1. 实验仪器与试剂•实验仪器:微量注射器、微量分析天平、恒温箱、紫外分光光度计、扫描电子显微镜(SEM)等。
•试剂:二甲苯、氯仿、甲醇、硝酸银、硫酸铜、麦角涨素等。
2. 样品制备黄精及其炮制品纤维素部分切成薄片,大小为 4 cm × 2 cm。
用短时间的微波处理干燥,然后用冷开水浸润片面 3-5 min,抹去水分,晾干后再切为 2 cm × 1 cm的小片。
3. 薄层色谱条件取硅胶G薄层板,均匀黏贴并通过紫外灯进行预处理(254 nm),然后将处理后的黄精薄片在薄层板上均匀均匀涂布后,将之竖直于气相色谱柱的溶解剂Toluene,四氯化碳,甲酸,甲醇,氯仿和乙醇的液体表面上。
薄层板通过气相色谱柱逆流扫描的方法,分别观察样品梯度染色、Visible light-UV 2D色谱图色标,UV 2D跳跃扫描等阳极和阴极图谱,最后用紫外光照射,然后将数据记录下来。
4. 鉴别方法1.薄层分析法:黄精薄层上普遍限定一种表征性的杂质鉴别方法——虎杖(角信)根薄层分析法:取黄精薄片与虎杖(角信)根薄层板进行比色分析,结合比色图谱,可以鉴别出黄精中的虎杖(角信)根。
2.假单宁试验:黄精中的黄酮类化合物可以在酸催化下缩合形成假单宁。
将黄精粉末加入硫酸乙酰去水溶液,再用加入氢氧化钠溶液时判断样品表面的颜色变化。
如果颜色从灰绿色变为深红色则为假单宁试验的阳性反应,证实了黄精中存在黄酮类化合物。
3.紫外吸收光谱分析:取黄精样品中的黄酮类成分,用75%乙酸溶液稀释,以 240-400 nm 范围内检测它的紫外吸收光谱,可以获得相应的峰值。
黄芪鉴定实验报告总结
本实验旨在对黄芪进行鉴定,并通过观察和实验结果对其进行总结。
黄芪是我国传统中药之一,具有很高的药用价值,然而市场上存在着一些假冒伪劣的黄芪产品,因此对黄芪进行准确的鉴定非常重要。
本实验采用了外观特征观察、显微镜观察和化学试验等方法对黄芪进行鉴定。
首先,通过外观特征观察,我们观察到黄芪的草本植物具有直立的茎,叶子互生,呈羽状复叶,花型为蝶形,花色为紫红色。
这些外观特征是黄芪的重要识别特征之一,并与文献上所描述的黄芪外观特征相符。
其次,通过显微镜观察,我们对黄芪的细胞结构进行了研究。
我们观察到黄芪的根部具有较长的细胞,细胞壁显著厚,而茎部和叶部的细胞较短而且较扁平。
这些细胞特征与文献所描述的黄芪的细胞结构相符,进一步表明了样本的真实性。
最后,我们进行了化学试验,采用了Iodine-Potassium Iodide
试剂和酸不水浸取液试剂。
结果显示黄芪与Iodine-Potassium Iodide试剂反应,呈现出紫褐色的沉淀,而未发现假阳性结果。
此外,酸不水浸取液试剂的化学导致剂也显示出了黄芪特有的化学反应。
综上所述,通过对黄芪的外观特征观察、显微镜观察和化学试验,我们对黄芪进行了准确的鉴定。
这些鉴定结果表明我们所用的黄芪样本是真实有效的,具有药用价值。
然后,我们也强
调了对黄芪进行鉴定的重要性,并提醒消费者在购买黄芪产品时要选择正规渠道,确保药材的质量和安全性。
第1篇一、实验目的通过对大黄药材的性状、显微特征、理化鉴别等方法进行综合分析,验证大黄药材的真伪,提高对大黄药材的鉴别能力。
二、实验材料1. 大黄药材:购自药材市场,分为疑似真品和疑似伪品两组。
2. 显微镜:光学显微镜及配套载物台、物镜、目镜等。
3. 显微镜切片机:用于制作大黄药材的横切片。
4. 紫外分光光度计:用于检测大黄药材中的大黄素含量。
5. 标准大黄药材:用于对照鉴别。
三、实验方法1. 性状鉴别观察疑似真品和疑似伪品大黄药材的外观特征,包括药材形状、颜色、气味等,与标准大黄药材进行对比。
2. 显微鉴别制作大黄药材的横切片,在显微镜下观察其组织构造,包括导管、纤维、筛管等细胞形态,与标准大黄药材进行对比。
3. 理化鉴别(1)大黄素含量测定:采用紫外分光光度法测定疑似真品和疑似伪品大黄药材中的大黄素含量,与标准大黄药材进行对比。
(2)薄层色谱法:将疑似真品和疑似伪品大黄药材进行提取,制备薄层色谱板,与标准大黄药材进行对比。
四、实验结果1. 性状鉴别疑似真品大黄药材呈类圆柱形,长3-10cm,直径1-3cm;表面黄棕色或暗棕色,有纵皱纹及网纹,有的可见横长皮孔;质坚实,断面黄棕色,颗粒性;气微香,味苦。
疑似伪品大黄药材呈不规则圆柱形,长3-7cm,直径1-2cm;表面黄褐色,有纵皱纹及网纹,有的可见横长皮孔;质较疏松,断面黄棕色,颗粒性;气微香,味苦。
与标准大黄药材外观特征基本一致。
2. 显微鉴别疑似真品大黄药材横切片在显微镜下可见导管为网状,纤维束与筛管群相间排列,筛管细胞呈类圆形或椭圆形,壁薄;疑似伪品大黄药材横切片在显微镜下可见导管为网状,纤维束与筛管群相间排列,筛管细胞呈类圆形或椭圆形,壁薄。
与标准大黄药材显微特征基本一致。
3. 理化鉴别(1)大黄素含量测定:疑似真品大黄药材中大黄素含量为1.25%,疑似伪品大黄药材中大黄素含量为0.8%,均高于国家标准(0.5%)。
与标准大黄药材大黄素含量基本一致。
中药分析重要知识点总结一、中药分析的原理中药分析的原理主要包括物理性质分析、化学成分分析和生物活性分析三个方面。
其中物理性质分析包括外观检查、气味、味道、溶解性等;化学成分分析包括定性分析和定量分析;生物活性分析则是评价药物的药效和毒性。
1.外观检查外观检查是中药分析的第一步,通过观察样品的形状、颜色、气味以及其他外在特征来初步判断样品的品质和真伪。
外观检查可以反映中药在保存过程中是否存在变质、虫蛀、霉变等情况。
2.物理性质分析物理性质分析包括测定中药的密度、溶解度、比旋光度、粒度分布等特性。
这些特性可以反映中药的物理性质和成分含量。
3.化学成分分析化学成分分析是中药分析的核心内容,包括定性鉴别和定量测定两个方面。
定性鉴别是通过化学反应、色谱色谱等手段判断中药样品的成分和含量;定量测定是测定中药样品中特定成分的含量,通常采用色谱法、质谱法、光谱法等。
4.生物活性分析生物活性分析是评价中药的药效和毒性的重要手段,包括细胞实验、动物实验和临床试验等方法。
生物活性分析可以反映中药的药效成分和毒性成分。
二、常用分析方法中药分析的常用分析方法主要包括色谱法、质谱法、光谱法和生物活性法等。
1.色谱法色谱法是中药分析的常用方法之一,包括气相色谱法、液相色谱法等。
色谱法可以分离和鉴定中药中的各种成分,是分析中药的重要手段。
2.质谱法质谱法是通过测定中药样品的质谱图谱来分析中药的成分和结构。
质谱法具有高灵敏度、高分辨率等优点,对中药的成分分析有很大的应用价值。
3.光谱法光谱法是通过测定中药样品的吸收光谱、荧光光谱、紫外光谱等来分析中药的成分和特性。
光谱法对中药的成分和性质分析有很大的应用价值。
4.生物活性法生物活性法是通过细胞实验、动物实验和临床试验等方法来评价中药的药效和毒性。
生物活性法可以直接反映中药的临床应用价值。
三、常见的分析技术和仪器中药分析常见的分析技术和仪器主要包括高效液相色谱仪(HPLC)、气相色谱仪(GC)、质谱仪(MS)、红外光谱仪(IR)、紫外可见分光光度计(UV-Vis)、原子吸收光谱仪(AAS)等。
浮选溶液中黄药及其分解产物的分析现状摘要:本文综述了国内外对选矿水溶液中黄原酸盐的分析测定方法。
阐述了紫外分光光度法、化学滴定法、气相色谱法、高效液相色谱法、毛细管电泳法的基本原理,优缺点及测定效果。
并指出黄原酸盐测试技术将向多种药剂同时测定的方向发展,对选矿厂药剂合理利用和分配具有重要的意义。
关键词:综述;黄原酸盐;测试技术;前言自1925年Keller首次在浮选过程中使用黄药作为捕收剂以来[1],关于黄药在浮选溶液中的变化规律,赋存状态的研究就倍受关注,因为这关系到黄药在浮选过程中的合理用量[2],自动化检测和控制,以及在选矿废水处理过程中具有重要的意义。
但由于矿浆中成分复杂,溶液中干扰因素多,黄药在浮选过程中分解产物繁多,仪器设备的限制,加上实验操作的精确性,使得分析过程难上加难,分析结果不尽人意。
近年来,随着现代科技的不断创新,技术的不断改进,对黄药及其衍生物测定的仪器方法更加精确和成熟。
本文总结了目前国内外对黄药及其衍生物在溶液中常用的测试手段,并对各方法进行了比较,对捕收剂等微量药剂在溶液中的测定具有重要的意义。
一紫外可见分光光度法紫外可见分光光度法在浮选研究中主要用于测定溶液中的低浓度浮选药剂,研究药剂与矿物作用产物的组成,某些调整剂在浮选过程中的作用,以及药剂吸附动力学等。
李文艳等[3]利用紫外可见分光光度计测定生产废水中乙基黄原酸钾的含量。
先过滤出废水中的不溶性物质后,以待测废水为背景样进行校正,直接测定吸光度,有效的消除了干扰,该方法检出限为0.01mg /L,方法的线性范围为0.04—18mg /L,水样测定的相对标准偏差为1.63%。
贺心然等[4]采用紫外分光光度(UV)法测定待测水样中丁基黄原酸浓度,用待测水样作为背景校正,并通过对不溶性物质,可溶性物质如硝酸盐,亚硫酸盐,以及金属离子的干扰实验,使得实际水样的测定相对标准偏差小于5.76%,检出限为0.006 mg/ L、测定上限为12.00 mg/ L,利用不同方法对样品进行分析测试,无明显差异。
F. Hao, K.J. Davey, W.J. Bruckard, J.T. Woodcock等[5]进行了黄药在实验室浮选过程中的在线监测研究。
通过紫外可见分光光度计等技术对黄药的浓度进行了实时测量,检出限达0.001mmol,利用HPLC对实时分析结果进行了比对,发现不同种类黄药的浓度与在线监测时相一致。
研究有利于在选矿厂在浮选过程中药剂制度的改善,从而节省药剂用量。
松全元[6]曾利用紫外光谱研究钛铁矿表面苄基胂酸吸附动力学曲线。
其将钛铁矿纯矿物置于数毫升苄基胂酸溶液中,在一定pH条件下搅拌后,溶液移入离心试管,离心分离得清液,用分光光度计测溶液的吸光度, ,即可求出钛铁矿吸附后苄基胂酸的剩余浓度。
由相应公式可算得矿物表面的吸附密度,从而得到吸附动力学曲线。
由于药剂吸附量一般在10-8一10-10mol/cm2数量级,这用常规分析方法是难以测定的,但利用紫外可见分光光度计通过测定与矿物作用后溶液中药剂浓度的变化,从而推算矿物表面的吸附量,是比较简单的。
余雪花等[7]研究了纯乙基黄药对纯黄铁矿相互作用行为的紫外光谱研究,在黄铁矿经硫酸铜活化后与黄药作用,除产生双黄药和一价铜的黄原酸盐外,还有二价铜的黄原酸盐,后者在高浓度的硫酸铜用量时较为显著。
同时研究了随着pH的增加,乙黄药与黄铁矿作用后产生的双黄药原来越少,pH达11.7时,双黄药的生成量为0,在碱性条件下pH>9时黄铁矿表面吸附的主要是另一种黄药的衍生物,即乙基一硫代碳酸盐,但它在黄铁矿表面会迅速分解。
利用紫外可见分光光度计进行各项测试,操作简单,方便,标准曲线线性相关性好,线性范围宽,是目前大家常用的测试手段。
但是只能同时测定单一组分,含有多种组分时,操作复杂,而且重复性较差。
二化学滴定法采用滴定法测定黄原酸盐是早期比较原始的分析方法,利用弱氧化剂进行标定,淀粉作为指示剂,当溶液由乳白色变成微黄色即达到终点。
主要有沉淀滴定法,硝酸银电位滴定法,示波滴定法等。
但该方法步骤繁琐,缺乏准确性,重复性差等缺点,目前仅适用常量分析。
陈景文等[8]采用丙酮分离试样中的共存物质,利用黄原酸于室温下,在酸性介质中定量分解生成CS2和相应的醇(合成反应的逆反应)的性质,采用酸碱滴定法测定碱金属黄原酸盐的含量,滴定终点敏锐,经与硝酸银电位滴定法,沉淀滴定法比较,本法操作简单,测定结果重现性好,相对标准偏差小于0.35%,可满足实际生产应用的需要。
王玉琴等[9]报道了以银电极为指示电极,饱和甘汞电极为参比电极,在丙酮—氨水介质中用硝酸银电位滴定法测定黄原酸钾的含量,滴定终点敏锐,方法简便,相对标准偏差低于0.5%,可满足实际生产应用的要求。
朱红霞等[10]通过对国家标准GB/T5750.08-2006中丁基黄原酸的测定方法进行了试剂的选择,pH影响,洗涤次数,静置时间,标准液体加入方式等条件的实验。
提高了方法的灵敏度,解决了实验过程中出现的常见的技术问题,应用该方法对饮用水及其水源水进行了测定,样品测定过程中发现该方法易受基体干扰,需要进一步探讨消除干扰的方法,拓宽该方法的适用范围。
三气相色谱法气相色谱法是二十世纪五十年代出现的一项重大科学技术成就。
它是指利用气体作为流动相的色谱法,当多组分的混合样品进入色谱柱后,柱内的吸附剂对各组分的吸附力不同,样品在流动相和固定相之间达到平衡,从而进行分离分析。
李冠军等[11]提出了应用气相色谱技术测定水中超痕量黄药的方法,其原理是根据黄药在强酸介质中定量分解出二硫化碳的特性,采用反推的方法求得黄药的浓度,作者采用顶空气相色谱技术测定超痕量黄药,特点是抽取与样品呈平衡状态的进行分析,根据气态方程与稀溶液特性求出黄药溶度。
该实验最低检测限达0.3ug/L,在含黄药5ug/L的水平其变异系数仅为5%。
张凯舟等[12]利用气相色谱法测定了微量黄药。
其利用黄原酸盐溶于水中电离出黄原酸根离子,根据物质在固定相和流动相之间的分配系数的不同,使得黄药阴离子与其他组分分离,分离后的组分经高灵敏性,高选择性的火焰光度检测器检测,使含硫原子的的物质产生一系列的特征反应,转变为光电流,经过放大记录,可以可获得定性定量的光谱图。
实验过程中采用担体为苯乙烯二乙烯苯的共聚物,柱温在160度,汽化室温度210度,检测器150度,一定的空气流速,氮气,氢气流速条件下,并使用8-羟基喹啉三氯甲烷为萃取液,消除了电解质溶液中离子的干扰。
实验结果表明黄药的浓度在0~50ug/mL的范围内与色谱峰峰高呈线性,加标回收率达94%以上。
气相色谱具有高分离效能,高选择性,高灵敏性的特点,并且操作简单,分析速度快。
但不能测定难挥发性,热稳定性不好的物质。
由于黄原酸盐受热易分解出CS2,在测定混合药剂时,不能分别测出不同烷基类黄药浓度。
四高效液相色谱法高效液相色谱法是在经典液相色谱之上引入气相色谱理论发展起来的,是目前应用最多的色谱分析方法。
原理是由于样品溶液中各组分在两相中具有不同的分配系数,在两相中经过反复多次的吸附- 解吸的分配,从而分离的过程。
周春山等[13]提出了三种适用于黄原酸盐混合物中单个黄原酸盐的分离和测定的高效液相色谱法,柱前氧化反相色谱法,柱前络合反相色谱法,柱前氧化正相色谱法。
同时还提出从黄原酸盐混合物复杂的色谱峰测定单个黄原酸盐的计算方法,使各个黄原酸盐的最低检测极限为0.15~2.0ng。
宁淑萍等[14]提出了高效液相色谱定性定量分析黄原酸盐与氨荒酸混合物组成的方法。
混合物经碘氧化后生成对称和非对称化合物,用正己烷萃入有机相, 在RP18柱上以甲醇-水为流动相,240nm处检测,其校准曲线的线性范围为6.2×10-5~2×10-3mol/L,平行测定7次,测试结果的RSD<2%。
F. Hao, K.J. Davey, W.J. Bruckard, J.T. Woodcock等[5]利用HPLC研究了不同黄药在浮选溶液中的分布情况。
作者从浮选槽中取出10ml矿浆,经过8um的针头过滤器得到澄清液,将50ul滤液注射进入HPLC中进行测定。
利用乙基黄药和戊基黄药在HPLC中的检出时间不同,从而将这两种黄药区分出来。
通过校正曲线可测得高达0.25mM乙基黄原酸钠,并且检出限可达0.001mM黄药。
高效液相色谱不受样品挥发度和热稳定性的限制,它非常适合分子量较大、难气化、不易挥发或对热敏感的物质、离子型化合物及高聚物的分离分析。
但其也存在一定的问题,如当将样品直接注入柱内时,柱内的压力上升,物质的保留时间会改变,从而影响分离效率,操作相对较难。
五毛细管电泳法毛细管电泳是以高压电场为驱动力,以毛细管为通道,根据样品中各组分之间淌度和(或)分配行为上的差异而实现分离的一种液相分配技术,它是经典电泳技术和现代微柱分离技术相结合的产物。
广泛应用于生命科学,生物技术,药物学医学等领域[15],应用在浮选药剂的检测方面很少,但实践证明其在检测黄药及多硫代碳酸盐含量时非常有效。
F. Hissner等[16]利用毛细管区带电泳同时检测不同烷烃黄药和磷酸酯。
在实验中,使用硼酸和硼砂作为缓冲溶液,氢氧化钠作为调整剂,电压为25kv时对水样进行分离测定,通过对比迁移时间和标准峰,检测出药剂种类和含量。
同时作者通过比较不同高压下注射和堆垛注射的注射方法,确定了堆垛注射能够提供最佳的灵敏度,并且能降低检出限至10-40mg/L。
TuomasSihvonen等[17]利用毛细管电泳仪在纯水和浮选水溶液中分离异丁基黄药和乙黄药,在纯水中异丁基黄药的检出限可达0.41mgL-1,异黄药的检出限可达0.025mgL-1,在浮选流程水中,检出限分别为0.62mgL-1和0.16mgL-1。
整个分析成分的过程所消耗的时间不超过10min。
J. Kemppinen, A. Aaltonen等[18]通过在线耦合毛细管电泳并联合紫外可见分光光度计检验的方法对选矿水溶液中的黄药进行了在线检测,研究了黄药的分解产物,尤其是硫代碳酸盐,硫代硫酸盐。
这种方法可以直接对各种类型的选矿厂废水进行测定而不需要进行样品的准备,只需在样品导入时外加加压注射器辅助,并且可以检测到10mgL-1以下的黄药,结果可靠,重复性高。
毛细管电泳法与高效液相电泳法相比,具有分析速度快,柱效高,前处理简单,洗脱液少,毛细管易清洗等特点,应用在浮选药剂方面非常有效。
但灵敏度及线性范围不如高效液相电泳仪,仅能实现微量制备等缺点。
六结论1.紫外可见分光光度法,气相色谱法虽然操作简单,方便,但由于不同黄药的最大吸收吸收峰相同,只能测出样品中黄药类的总含量,不能分别测出不同黄药的含量。