4-硝基-2,3,5-三甲基吡啶-N-氧化物的合成
- 格式:pdf
- 大小:168.93 KB
- 文档页数:4

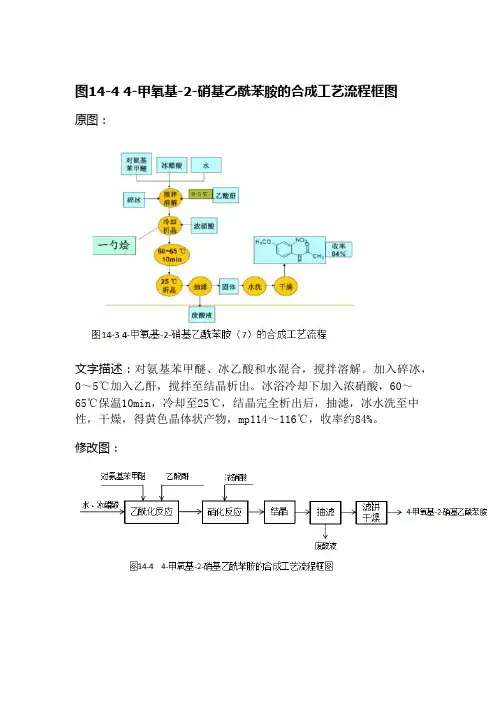
图14-4 4-甲氧基-2-硝基乙酰苯胺的合成工艺流程框图原图:文字描述:对氨基苯甲醚、冰乙酸和水混合,搅拌溶解。
加入碎冰,0~5℃加入乙酐,搅拌至结晶析出。
冰浴冷却下加入浓硝酸,60~65℃保温10min,冷却至25℃,结晶完全析出后,抽滤,冰水洗至中性,干燥,得黄色晶体状产物,mp114~116℃,收率约84%。
修改图:图14-5 4-甲氧基-2-硝基苯胺的合成工艺流程框图原图:文字描述:将4-甲氧基-2-硝基乙酰苯胺原料加入Claisen碱液中,加热回流15min,加水,再回流15min,冷却至0~5℃结晶,抽滤,冰水洗3次,得砖红色固体产物,mp122~123℃,收率约88%。
修改图:图14-6 4-甲氧基邻苯二胺的合成工艺流程框图原图:文字描述:SnCl2与浓盐酸混合溶解,20℃下加入4-甲氧基-2-硝基苯胺,搅拌反应3h。
滴加40%NaOH液至pH=14,控温不超过40℃。
用乙酸乙酯萃取2次,水洗有机相,无水Na2SO4干燥。
减压脱出溶剂,黄色油状物冷冻结晶,得产物4-甲氧基邻苯二胺,收率约72%。
修改图:图14-7 5-甲氧基-1H-苯并咪唑-2-硫醇制备工艺流程框图原图:文字描述:搅拌下将4-甲氧基邻苯二胺和CS2加到95%EtOH和KOH的混合液中,加热回流3h。
加入活性炭回流,趁热过滤。
滤液搅拌下滴加乙酸至pH=4~5析出结晶,冷却至4~5℃使析出完全。
抽滤,水洗至中性,干燥,得土黄色产物结晶,mp254~256℃,收率约78%。
修改图:图14-8 2,3,5-三甲基吡啶-N-氧化物的制备工艺流程框图原图:文字描述:将2,3,5-三甲基吡啶与H2O2、HAc混合,搅拌下缓缓升温至80~90℃,反应24h。
减压蒸除溶剂,冷却,用40%的NaOH调节pH =14,用CHCl3萃取3次,无水Na2SO4干燥。
减压浓缩,50~60℃真空干燥,得黄色固体产物,收率80.3%。
修改图:图14-9 4-硝基-2,3,5-三甲基吡啶-N-氧化物制备的工艺流程框图原图:文字描述:搅拌、控温<90℃,将硫酸滴加到三甲基吡啶氧化物中,缓慢滴加混酸(硫硝比=1∶1.10),90℃保温反应20h。



奥美拉唑生产工艺规程编号:SOP-MF-301-19起草:日期:审阅:日期:审核:日期:审批:日期:执行日期:分发部门:生产部,设备部、办公室、质检部目录1.产品概述 (1)1.1通用名称 (1)1.2化学名 (1)1.3中文别名 (1)1.4汉语拼音 (1)1.5英文名称 (1)1.6常见的商品名 (1)1.7英文别称 (1)1.8分子式 (1)1.9分子量 (1)1.10结构式 (1)1.11理化性质 (1)1.12执行的标准 (1)1.13临床用途 (1)2.产品包装及规格 (1)2.1包装 (1)2.2包装规格 (1)2.3规格 (1)2.4储存 (1)3.原辅料、包装材料质量标准 (1)3.1原辅料、成品质量标准 (2)3.2包装材料 (4)3.3说明书 (4)4.化学反应式与工艺流程图 (4)4.1化反应式 (4)4.2生产流程图 (6)4.2.1工艺流程方框图 (6)4.4.2设备流程图 (9)5.工艺过程 (9)5.1配料比 (9)5.2操作工艺过程 (10)5.3重点工艺控制点 (11)5.4异常情况处理 (11)5.5注意事项 (11)6.中间体、半成品质量标准 (12)6.1中间体的质量标准 (12)6.2半成品布洛芬的质量标准 (12)7.技术安全与防火 (12)7.1技术安全 (12)7.2劳动保护与卫生安全 (13)7.3洁净区环境监测要求 (13)7.3危险化学品一览表 (13)8.综合利用与三废治理 (18)8.1.综合利用 (18)8.2三废治理 (18)9.操作工时与生产周期 (18)10.劳动组织与岗位定员 (19)11.设备一览表及主要设备生产能力 (19)11.1设备一览表 (19)11.2主要设备生产能力 (20)12.原材料、能源消耗定额和技术经济指标 (20)12.1能源消耗定额 (20)12.2技术经济指标 (20)13.物料平衡 (20)14.附录 (22)14.1物料平衡计算公式 (23)14.2换算表 (23)15.附页 (23)1.1 产品名称:【中文名称】:奥美拉唑【简写拼音】: AMLZ【外文名】Omeprazole、Losec【化学名称】5-甲氧基-2-{[(4-甲氧基-3,5-二甲基-2-吡啶基)-甲基]-亚磺酰基}-1H-苯并咪唑1.2产品【结构式】【分子式】C17H19N3O3S【分子式量】345.421.3理化性质:本品为白色或类白色结晶性粉末;无臭;遇光易变色。

几类N-氧化物的应用和直接合成方法易亮;孙德群【摘要】N-氧化物是一类具有广泛生物活性的化合物,常用于各种药物合成的中间体或者本身就是药物;同时它们也可以作为有机化工中间体和催化剂等,在有机合成上起到非常重要的作用.传统的制备N-氧化物的方法是采用合适的氧化剂对含氮化合物进行N-氧化.此外,用直接合成方法,即在生成含氮化合物的同时发生N-氧化,也可以得到相应的N-氧化物.由于直接合成方法的一些优势和传统方法的诸多缺陷,本文着重总结了包括嘧啶类N-氧化物和喹喔啉类N-氧化物等在内的十类N-氧化物的直接合成方法,并简述了它们的应用.【期刊名称】《四川大学学报(自然科学版)》【年(卷),期】2016(053)003【总页数】20页(P606-625)【关键词】N-氧化物;药物;有机合成;直接合成方法【作者】易亮;孙德群【作者单位】山东大学(威海)海洋学院,威海 264209;山东大学(威海)海洋学院,威海264209【正文语种】中文【中图分类】O623.7;O626N-氧化物因含有氮氧偶极结构而使其具有独特的性质.它们通常具有广泛的生物活性,广泛应用于医药[1-3]和农药[4-7]等领域.此外,它们也是很多生物活性化合物[8, 9]和手性配体[10, 11]的结构组成单元.在有机合成上,它们也通常被用作合成复杂分子[12, 13]和天然产物[14, 15]的中间体.得到N-氧化物的传统方法是采用合适的氧化剂,对含氮化合物进行N-氧化.此外,用直接合成方法,即选用合适的底物在一定条件下反应,在生成含氮化合物的同时发生N-氧化,也能够得到相应的N-氧化物.由于传统方法存在诸多不足,一是在合成含氮杂环化N-氧化物时,需要提前提供含氮杂环化合物[16];二是氧化剂在氧化含有多个氮原子或是存在易氧化的结构的含氮化合物时,存在氧化选择性问题[16, 17];三是随着含氮原子数目的增加,进行N-氧化反应的难度增加,对氧化剂和反应条件的要求也更加苛刻[18].而对于直接合成方法来说,由于它是在合成含氮化合物的同时发生N-氧化,因此节省了反应的步骤,使产物收率较高,且通常不存在采用传统方法所带来的问题.另外,当需要在含氮杂环N-氧化物上引进一些官能团时,可以通过直接合成方法,采用含有相应官能团的底物来引进,而且生成的N-氧化物因含有丰富的功能结构而使其能够进一步与其他化合物反应,生成各种相应的N-氧化物衍生物,这些衍生物(或其去氧化后转换成相应的含氮化合物)通常是一类具有生物活性的化合物[12-15, 19-21].但是目前这种方法只适用于部分N-氧化物的合成,因此不够普遍[18].不过随着合成方法的不断发展,越来越多的N-氧化物通过直接合成方法被合成得到.鉴于N-氧化物在诸多领域中的应用,因此合成此类化合物具有重大的意义.综合考虑传统方法的诸多缺陷和直接合成方法的一些优势,本文选用了十类N-氧化物,对合成这些N-氧化物的直接合成方法方法作了较为全面的总结,并简述了这十类N-氧化物的相关应用.嘧啶类N-氧化物是一类多功能化合物.它们既具有广泛的生物活性,又可以在有机合成中被用作中间体.如嘧啶类N-氧化物米诺地尔就是一个高效的促进血管舒张和抗高血压的分子[22].另外,它们也被用作生长调节剂和除草剂[7]以及抗皮炎[23]和预防脱发[24]的药物.在有机合成上,如2-氨基嘧啶类N-氧化物作为中间体被广泛应用于合成其他杂环体系[25].Buscemi等[26]采用3-氨基-5-甲基-1,2,4-恶二唑1和β-二酮类化合物2在高氯酸条件下反应,得到相应的嘧啶类N-氧化物6(图式1).在这个环到环的转化过程中,先是1和2发生亲核加成反应,得到加成物3,然后3经历分子内缩合环化脱水,得到1,2,4-恶二唑嘧啶盐4,紧接着4发生水解作用,在恶二唑部分发生开环,得到5,5再进一步水解,得到终产物6.反应式为:在这个反应中,当用2a反应时,产物收率只有21%,且会发生各种副反应;当用2b反应时,产物收率为50%,也会发生与2a相同的副反应;而当用2c反应时,产物收率高达92%,且不发生副反应.Murashima等[27]采用硝基杂芳族类化合物与异腈基乙酸乙酯在DBU(1,8-二氮杂二环十一碳-7-烯)存在条件下,反应得到相应的嘧啶类N-氧化物或吡咯类化合物.如用5-硝基-2,1,3-苯并噻/硒二唑7与异腈基乙酸乙酯反应,得到相应的嘧啶类N-氧化物8(图式2).反应式为:在这个反应中,7的硝基在芳环上取代的位置不同,将生成不同的产物.如当底物为4-硝基-2,1,3-苯并噻/硒二唑9时,则生成单一的吡咯类化合物.这主要取决于硝基与芳环是否共平面,若共平面则生成相应的嘧啶类N-氧化物,反之则生成相应的吡咯类化合物.Jalli等[28]采用4-氯-3-甲酰香豆素10和芳香族甲酰亚胺肟类化合物11通过双亲核加成串联和脱水反应,得到相应的苯并吡喃嘧啶类N-氧化物12(图式3).反应式为:在这个反应中,当R为卤素或供电子取代基时,得到收率很高的卤素或供电子取代的嘧啶类N-氧化物;当R为吸电子取代基硝基时,在该反应条件下没有得到硝基取代的12,将反应溶剂换成1,4二恶烷,并提高反应温度,得到了收率较好的硝基取代的12.另外,由于脂肪族类甲酰胺肟在该实验条件下不稳定,因此反应底物中的肟类化合物必须是芳香族类甲酰胺肟.喹喔啉类N-氧化物根据其N-氧化的个数,分为喹喔啉类单N-氧化物和双N-氧化物.它们都具有非常广泛的生物活性.对于单N-氧化物来说,它们具有抗感染[29-31]、抗癌[32]以及抗结核分枝杆菌[33]的活性,同时也被用作血管紧张素Ⅱ受体的拮抗剂[34].对于双N-氧化物来说,它们具有抗结核分枝杆菌[35]、抗疟疾[36]、抗锥体虫[37]以及抗癌[38, 39]等活性.而且由于它们允许官能团和结构的修饰,使其具有重大的合成价值[20, 40, 41].Tennant等[17]采用α-苯甲酰邻硝基乙酰苯胺13与正碘烷(C2 ~ C7)在NaCO3溶液中进行烷基化,生成C-烷基化的α-苯甲酰邻硝基乙酰苯胺14在热碱溶液中进行环化,得到相应的氧喹喔啉类单N-氧化物15(图式4).反应式为:在这个反应中,两步反应的收率都很高,但用二级或三级碘烷不能对13进行烷基化.Takano等[20]采用4-氟-2-硝基苯胺16与乙基丙二酰氯17在Et3N/DMF中进行缩合反应,得到缩合物18,紧接着18在tBu-K/EtOH中进行分子内的环化,得到相应的喹喔啉类单N-氧化物19(图示5).反应式为:Monge等[42]采用苯并氧化呋咱20与烯胺类化合物21在CH3COONa溶液中反应,得到收率小于35%的喹喔啉类双N-氧化物22(图式6).反应式为:在这个反应中,当把21换成炔类化合物RC≡CH(R=CH2OH,Ph ,-CO2H)时,在含有Et3N,Et2NH或BuNH2的异丙醇中与20反应,也将得到相应的喹喔啉类双N-氧化物.Issidorides等[43]采用苯并氧化呋咱23与1,3二酮类化合物或β-酮酯类化合物24在碱性条件下反应,得到相应的喹喔啉类双N-氧化物25(图式7).反应式为:在这个反应中,当碱为Et3N时,23分别与24a,24b,24c反应,对应生成25a,25b,25c;当用NaOH/EtOH替代Et3N作碱时,24a与23反应得到收率为90%的25a的脱酰基产物25.24b和24c分别与23反应,则没有像24a那样生成相应的脱酰基产物,而是生成了一些难以处理的混合物.这是由于在2,3位烷基取代的喹喔啉类双N-氧化物在强碱条件下不稳定的原因造成的.Abushanab等[44]采用邻苯醌二肟26和α-二酮类或α-羟基醛类化合物27共热,得到相应的喹喔啉类双N-氧化物28(图式8),产物收率较低.反应式为:在这个反应中,对于α-羟基醛或酮来说,首先是26的一个肟基对27的羰基进行亲核加成,而26的另一个肟基互变异构为亚硝基,将27的羟基氧化成羰基,自身被还原为羟胺,然后羟胺与羰基进行亲核加成,发生分子内环化,脱水得终产物28.对于α-二酮类化合物来说,首先是26和27发生氧化还原反应,26被氧化成苯并氧化呋咱,27被还原成α-羟基醛或酮,然后被还原的27再与另一分子的26进行前述反应.Meyer等[45]采用1,2,2-三氯-2-硝基乙烯(TCNiE) 29与苯胺以1:1的比例,在40℃的Et3N/MeOH中反应,得到相应的喹喔啉类单N-氧化物31(图式9).反应式为:在这个反应中,先是29与苯胺发生Michael加成,得到加成物32,然后32的胺盐基上的质子转移至硝基的氧上,得到中间体30的等价物氮酸33,33发生分子内环化脱水后,得到34,最后34水解生成终产物31.另外,苯环上含有取代基的苯胺也能发生该反应.Aggarwal等[46]采用多种合适的取代苯胺类化合物35和(E)-苯偶酰单肟36在EtOH/H2O中反应,得到各种收率的苯偶酰-α-芳亚胺基肟类化合物37,然后34在二乙酸碘苯(IBD)的氧化作用下,进行分子内环化,得到收率为60-75%的喹喔啉类单N-氧化物38(图式10).反应式为:在这个反应中,所用的氧化剂IBD相比于用Pb(OAc)4作氧化剂来说,反应更加温和安全,产物的收率也更高.Krolikiewicz等[47]采用N-芳香胺类化合物与芳硝基类化合物反应所得到的N-芳基-2-亚硝基苯胺类化合物39与氰基乙酸酯的烷基化衍生物40在碱性条件下反应,得到相应的喹喔啉类单N-氧化物41(图式11),产物收率为48-96%.反应式为:在这个反应中,首先是41的碳负离子和39的亚硝基缩合,得到硝酮中间体,紧接着发生氨基的分子内酰化,得到终产物41.该反应一般用DBU/MeCN作为碱体系,但在有些情况下,用K2CO3/DMF作为碱体系时,产物收率更高.Chen等[16]开辟了一条合成喹喔啉类N-氧化物的新方法,通过采用各种胺类化合物和不同的芳香酮类化合物反应所得到的不同取代的亚胺类化合物42为底物,与亚硝酸叔丁酯(TBN)43反应,得到相应的喹喔啉类单N-氧化物44(图式12),产物收率较好.反应式为:在这个反应中,43作为NO的来源,反应过程经历三个C-H键的消除和C-N键形成,反应机理为自由基反应过程,且不需要过渡金属催化.另外,四丁基溴化铵(TABA)的加入有利于稳定NO自由基,使其浓度增加,从而提高反应收率.硝酮根据其结构是否成环,分为环硝酮和非环硝酮.它们既具有广泛的生物活性,同时也是一类重要的有机合成中间体.常常被用作自由基自旋捕捉剂[3]和用于治疗神经衰弱、心血管疾病和癌症等氧化应激相关的疾病[48-50].如环硝酮45(DMPO)是一种最常用的自由基自旋捕捉剂[51],而非环硝酮46,可以用来治疗急性中风[52],同时还在胶质瘤模型[53]和肝癌细胞[54]中表现出抗癌活性(图式13).另外,由于它们既可以作为1,3偶极体和亲偶极体进行1,3偶极环加成[55],又可以作为亲电体进攻有机金属配合物[56, 57],因此在合成上起到非常广泛的作用.目前合成硝酮的一般方法是羟胺类化合物与羰基类化合物缩合得到[58].Bonnett 等[59]在合成环硝酮吡咯啉类N-氧化物时,采用γ-硝基-醛/酮类化合物47在Zn 粉和冷的NH4Cl溶液中还原,得到γ-羟胺醛或酮类化合物48,然后48再进行分子内环化,得到相应环硝酮吡咯啉类N-氧化物49(图式14).反应式为:在这个反应中,47的羰基有可能被还原,必要时需要先对其进行保护,最后水解得到终产物49.Karoui等[60]采用硝基丙二酸二乙酯50与丙烯醛51反应,得到环硝酮二乙氧基羰基-1-吡咯啉N-氧化物53(DECPO)(图式15).反应式为:在这个反应中,50和51先经历Michael加成反应,得到加成物52,然后52的硝基在Zn/NH4Cl作用下,还原成羟胺,紧接着与羰基进行缩合环化,得到终产物53.Lebel等[61]采用醛/酮肟O-三甲基硅醚类化合物54与三烷基氧鎓四氟硼酸盐/烷基三氟甲磺酸在室温或低于室温的条件下反应,得到收率较好的非环硝酮类化合物57(图式16).反应式为:在这个反应中,首先是三烷基氧鎓四氟硼酸盐或烷基三氟甲磺酸对54进行N-烷基化,得到N-烷基化盐中间体55,紧接着55脱甲硅基,得到BF3复合物56,由于56一般比较稳定,因此需要向其加入含有氟化钾的乙腈溶液,再进行过滤,蒸发,结晶或蒸馏等操作,得到终产物57.Tufariello等[62]采用异恶唑类化合物的热裂环(thermal cycloreversion)反应,得到相应的环硝酮类化合物.例如异恶唑类化合物58在m-CPBA氧化作用下开环,得到相应的环硝酮59(图式17),收率为89%.反应式为:在这个反应中,59在无机酸作用下或在含有对甲基苯磺酸的苯溶液中回流,均不发生脱水作用.Lebel等[63]采用N-羟胺基磺酸盐类化合物60的Grob断裂(Grob fragmentation)反应,得到相应的环硝酮类化合物61(图式18).反应式为:在这个反应中,61的收率与碱的强度有关,如当碱为100%的乙醇时,则主要发生对甲苯磺酸根(TsO-)与相邻碳上氢的E1消除反应,当碱为氢化钠或叔丁醇钾等强碱时,则主要发生Grob断裂反应,生成终产物61.跟其它叔胺类N-氧化物一样,简单的烯胺类N-氧化物具有吸湿性.它们在有机合成上可用作合成中间体,如Hwu等[64]采用其作为合成吲哚类化合物的中间体.另外,它们还具有一些其他合成作用,如奎宁环核是抗疟疾活性化合物金鸡纳碱的核心单元,其N-氧化物是一种烯胺N-氧化物,它可以替代六甲基磷酰三胺在很多重要合成反应中所起的作用[65],且其二聚物具有增强金属结合的性质[66].此外,它们还有很多理论上的应用尚待进一步的研究[67].Winterfeldt等[68]采用二乙基羟胺62和二甲基乙炔二羧酸酯63进行分子间的reverse-cope环加成反应,得到不稳定的烯胺类N-氧化物64(图式19).反应式为:Padwa等[69]采用初级羟胺与活化的炔类化合物反应,生成的终产物异恶唑类化合物是由该反应生成的中间体烯胺N-氧化物经异构化得到相应的硝酮,再与活化的炔进行1,3偶极环加成得到的.Krenske等[67]也报道了采用初级羟胺类化合物和炔类化合物生成的中间体烯胺类N-氧化物来合成生物碱.如用一级羟胺类化合物65与乙炔66反应,得到中间体烯胺类N-氧化物67(图式20).反应式为:基于这类合成方法,O'neil等[70]采用手性脯氨羟胺衍生物68与活化的炔类化合物69进行分子间的reverse-cope环加成反应,得到稳定单一的非对映异构的烯胺类N-氧化物70(图式21),产物收率很高.反应式为:在这个反应中,若69的一端为活化基团(如:-COOCH3)取代,而另一端为烷基、芳基或甲硅烷基取代,将不能与68发生该反应.腈类N-氧化物是一类重要的有机合成中间体.比如它们作为一种1,3偶极体,能够和烯烃Maugein等[73]采用2-苯基-1-硝基乙烷71在各种脱水试剂作用下脱水,生成相应的腈类N-氧化物72(图式22).反应式为:Edward等[75]在采用苯基硝基甲烷73在不同酸度的酸催化作用下研究Meyer反应机理时发现:在高酸度时,反应生成中间体腈类N-氧化物77(图式23).反应式为:在这个反应中,先是73的酸式结构74在酸催化作用下,生成75,然后75在与其N相连的两个羟基之间脱水,得76,最后76去质子化得到终产物77.Grundmann等[76]采用醛肟类化合物与卤化试剂反应,生成的肟基卤化物在碱诱导下脱去卤化氢,生成相应的腈类N-氧化物.如用醛肟类化合物78与卤化试剂反应,得到腈类N-氧化物79(图式24).反应式为:在这个反应中,先是NBS对78进行溴化,然后再脱去溴化氢得到79,产物收率高达90%.Yao等[77]采用β-硝基苯乙烯类化合物80a和80b分别与格氏试剂或烷基锂试剂81反应,在生成的中间体82中,慢慢加入浓度为85%的硫酸,最后生成腈类N-氧化物83(图式25).反应式为:在这个反应中,当80a进行该反应时,生成的中间体在不同的溶剂中反应,产物将出现三种情况:1)只生成腈类N-氧化物;2)只生成肟基类卤化物;3)两者都有.且由80a生成的腈类N-氧化物因含有两个苯环,使其有足够大的位阻而不被水解.当80b进行该反应时,只生成相应的羧酸,这是由于80b形成的腈类N-氧化物或肟基类卤化物的位阻太小而直接被水解成相应的羧酸.在Yao等[77]的方法上略加修改,Wang等[74]采用1,1-二苯基硝基乙烯84与正丁基锂反应,得到相应的腈N-氧化物85(图式26),产物收率高达95%.反应式为:在这个反应中,首先是正丁基锂与84进行Michael加成,紧接着在浓硫酸作用下脱水得到85.由于85连有大的取代基,可以阻止自身发生二聚作用,因此能足够稳定的被分离得到.在这个反应的基础上,他们又采用乙烯基单体的阴离子聚合物88来合成聚合腈类N-氧化物89(图式27).反应式为:在这个反应中,先是用仲丁基锂和二苯乙烯组成的混合物87在-78℃条件下,作为乙烯基单体86聚合的引发剂,得到其聚合物后,再加入1,1-二苯基硝基乙烯与其反应,得到的混合物在浓硫酸脱水作用下生成89.它是一种直接嫁接试剂,能与含有不饱和键的聚合物进行有效地点击嫁接[(2+3)环加成],在无溶剂和催化剂条件下得到转化率很高的各种高度复杂的接枝共聚物.吲哚酮类N-氧化物经常被报道其具有多种生物活性,例如它们能够抑制哺乳类动物线粒体中的ATP合成[78].在体外实验中,它们还表现出抗细菌[79]和抗真菌的活性[80].引起平滑肌放松[81]和保护神经的作用[82].另外,由于其含有硝酮的结构,因而也能够作为自由基自旋捕捉剂捕捉羟基自由基和过氧化物自由基[83].近年来,很多研究表明一些吲哚酮类N-氧化物还具有抗疟疾的活性[2, 84, 85].Genisson等[86]采用邻硝基苯二酮衍生物92合成得到了相应的吲哚酮类N-氧化物93(图式28).反应式为:在这个反应中,先是由90发生Wittig反应得到91,91再被KMnO4/醋酐氧化得到92,紧接着 92再在THF/CH2Cl2溶液中被Zn/NH4Cl还原,加热回流后发生分子内环化,得到93,收率为35-91%.通常步骤ⅱ需要1-2周的反应时间.Basavaiah等[87]研究了用苯亚硒酸酐(BSA)替代ⅱ中的KMnO4/醋酐,将烯烃类化合物氧化成1,2-二酮类化合物.研究表明:当R2=Aryl时,产物收率较高,而当R2=Alkyl时,效果不甚理想,并且认为在合成芳基,烷基-二酮类化合物时,KMnO4/醋酐是最佳的氧化剂.Nepveu等[85]在研究吲哚酮类N-氧化物的抗疟疾活性中,采用一种基于邻碘/溴硝基芳烃类化合物96与末端炔类化合物97的Sonogashira偶合反应,得到中间体98,然后98在一定的反应条件下发生分子内环化,生成相应的吲哚酮类N-氧化物99(图式29).反应式为:在这个反应中,97是94经反应ⅰ得到95,95再经反应ⅱ得到的.整个反应的产物收率仅有20-30%,但较于Genisson等[86]的方法来说,反应时间大大缩短,仅需0.5-17h,不过对于比较稳定的中间体98来说,由于它不能进行分子内环化而使反应停留在反应ⅲ,这时就需要用Genisson等[86]的方法来合成相应的吲哚酮类N-氧化物.Najahi等[84]也用了类似的方法合成了一些双吲哚酮类N-氧化物,用于研究其抗疟疾活性.吡啶类N-氧化物具有非常广泛的生物活性,它的结构出现在很多药物和具有生物活性的化合物中[8],同时也是一种有机催化剂,如Malkov等[88]用联吡啶双N-氧化物作为手性有机催化剂催化芳香醛的不对称烯丙基化.并且能够和某些有机金属形成配合物[89],例如吡啶类N-氧化物缩氨基硫脲2-取代吡啶氮氧化物的铜配合物是一种抗肿瘤试剂,在淋巴系统白血病细胞中,铜配合物优先抑制病菌DNA 合成[1].Crabbe等[90]采用4-亚丙酮基-2,6-二甲基-4-氢吡喃100与NH2OH·HCl反应,得到相应的吡啶类N-氧化物101(图式30).反应式为:Nakamura等[91]采用(E)-O-炔α,β-不饱和肟类化合物102在CuBr(PPh3)/PPh3催化作用下,得到相应的吡啶类N-氧化物103(图式31),产物收率很高.反应式为:在这个反应中,先是一分子102的氮原子对另一分子102的碳碳三键在铜催化作用下进行亲核加成,得到环化中间体104,然后104经历C-O键的异裂及铜催化剂的消除,得到N-丙二烯基硝酮中间体105,105的旋转异构体106再经历6π-3-氮杂三烯的电环化,得到二氢吡啶类化合物107,最后107异构成终产物103.Shi等[92]采用α,β-不饱和肟类化合物108和重氮类化合物109作为起始原料,合成得到相应的吡啶类N-氧化物110(图式32).反应式为:在这个反应中,先是Rh(Ⅲ)催化活化108的乙烯基的C-H键,然后再与109提供的卡宾串联形成C-C键,最后经历环化和缩合过程,得到110,产物收率很高.反应条件温和,不需要氧化剂,副产物为氮气和水.异喹啉类N-氧化物是一类应用非常广泛的化合物[93].常常被用来合成具有生物活性的化合物以及其他功能物质,如它们是合成异喹啉及其衍生物的中间体[94, 95].近年来,它们在不对称转化上被用作手性配体的骨架结构,并合成了一些新的化合物[96].由于它们在很多领域都有重要的用途,因此是一类非常重要的化合物. Wang等[97]报道了一种在水溶液中有效合成各种异喹啉类N-氧化物的方法,采用2-炔苯甲醛肟类化合物111和在原位产生的次溴酸盐在水溶液中进行亲电环化,得到相应的异喹啉类N-氧化物112,反应条件温和,产物收率也很高.反应式为:在这个反应中,次溴酸盐和次碘酸盐是由过硫酸氢钾分别和溴化钠和碘化钠在温和的条件下生成的,虽然卤素是一个有效地亲电试剂,但是由于它的毒性及易挥发性,所以用过硫酸氢钠-溴化钠的组合代替溴,简化了步骤并降低了对操作人员和环境的毒害.Ding等[98]用一锅煮的方法,采用2-炔苯甲醛肟类化合物113与卤代芳烃在Ag和Pd催化作用下合成得到了1-位芳化的1,3-二取代异喹啉类N-氧化物115.反应式为:在这个反应中,首先是113在Ag催化作用下,发生分子内加成-环化,得到114,然后再与卤代芳烃在Pd催化作用下直接芳化,合成得到终产物115,产物收率很高.在该反应条件下,114能够和各种取代的卤代芳烃进行芳基化反应,但当用[(E)-(2-溴乙烯基)苯]与114反应时,没有得到相应的产物.Huo等[99]采用2-炔苯甲醛肟类化合物116在碘介导的乙醇溶液中进行环化,合成得到3,4-二取代碘异喹啉N-氧化物117(图式35),产物收率很高.反应式为:在这个反应中,首先是116的碳碳三键在碘鎓阳离子的协调作用下被活化,然后其对肟氮进行亲核进攻,得到碘异喹啉中间体,紧接着去质子化得到117.该反应条件温和,底物官能团的耐受性也很好.Tamura等[100]建立了另一种合成异喹啉N-氧化物的方法(图式36).反应式为:在这个反应中,先是用等摩尔的异喹啉和2,4-二硝基氯苯混合物在40℃条件下反应5h得到定量的N-(2,4-二硝基苯)异喹啉氯化物118.A路线:在室温条件下,加入盐酸羟胺与119在Et3N/CH3OH中反应,得到收率为71%的121.B路线:先用5%的NaCO3在水溶液中与118反应生成高收率的119,然后将119与盐酸羟胺在Et3N/MeOH中反应,转化为121,总收率为68%.C路线:将119在20%的丙酮溶液中回流得到120,再将120与盐酸羟胺在Et3N中反应,得到121,从118到121的总收率为70%.由此可见A路线是最佳的.然后将121在各种溶剂(HCl-EtOH,HCl,AcOH,TsOH-EtOH,dioxane-H2O)中加热环化,得到终产物122.结果表明在HCl-EtOH溶剂中得到产物的收率最高(75%).Yoshida等[101]采用酮肟类化合物123与炔类化合物124在Ni催化作用下,得到3,4-二取代异喹啉类N-氧化物125(图式37),产物收率适中.反应式为:在这个反应中,在生成125的同时,也会伴随有相应的异喹啉类化合物的形成.当。

奥美拉唑的生产工艺原理班级制药10-4学号201004021021姓名赵成刚一、概述:【中文名称】:奥美拉唑【别名】:安胃哌唑;奥美拉唑;奥咪拉唑;甲氧磺唑;沃必唑;渥米哌唑;亚枫咪唑,洛赛克【外文名】:Omeprazole、Losec【化学名称】:5-甲氧基-2-{[(4-甲氧基-3,5-二甲基-2-吡啶基)-甲基]-亚磺酰基}-1H-苯并咪唑【结构式】:H3CONN H SONCH3OCH3H3C【理化性质】:为白色至类白色结晶性粉末,熔点156℃。
溶于二氯甲烷,微溶于水,乙醇和甲醇,易溶于氢氧化钠和氢氧化钾稀水溶液。
在276nm和305nm 的波长处有最大吸收(0.1mol/L氢氧化钠溶液,20ug/mL)。
奥美拉唑与华法林相互作用,可诱发细胞色素P450活性增强,而使血清胃泌素水平增高。
同时有研究证实本品不影响血浆阿司匹林和水杨酸浓度,20mg/d,在人体中不干扰阿司匹林对血小板的生物活性。
【药理作用】:选择性性地作用于胃粘膜壁细胞,抑制处于胃壁细胞顶端膜构成的分泌性微管和胞浆内的管状泡上的H+,K+-ATP酶的活性,从而有效地抑制胃酸的分泌,起效迅速,适用于胃及十二指肠溃疡,返流性食管炎和胃泌素瘤。
这种H+,K+-ATP酶抑制剂又名质子泵抑制剂。
由于H+、K+-ATP酶是壁细胞泌酸的最后一个过程,故本品抑酸能力强大。
它不仅能非竞争性抑制促胃液素、组胺、胆碱及食物、刺激迷走神经等引起的胃酸分泌,而且能抑制不受胆碱或H2受体阻断剂影响的部分基础胃酸分泌,对H2受体拮抗剂不能抑制的由二丁基环腺苷酸(DcAMP)刺激引起的胃酸分泌也有强而持久的抑制作用。
本品对胃蛋白酶分泌也有抑制作用,对胃黏膜血流量改变不明显,也不影响体温、胃腔温度、动脉血压、静脉血红蛋白、动脉氧分压、二氧化碳分压及动脉血pH。
二、研究进展:目前质子泵抑制剂主要有ATP-拮抗剂和K+-拮抗剂两类,ATP-拮抗剂为不可逆PPI, K+-拮抗剂为可逆PPI。



奥美拉唑的合成
万欢;方峰;段梅莉;许煦;冀亚飞
【期刊名称】《应用化学》
【年(卷),期】2009(26)2
【摘要】由2,3,5-三甲基吡啶经氧化、硝化一锅煮方法制得2,3,5-三甲基-4-硝基吡啶-N-氧化物. 该产物与三氯异氰尿酸直接进行氯化反应得到关键中间体2-氯甲基-3,5-二甲基-4-硝基吡啶-N-氧化物,在过量的甲醇钠存在下与2-巯基-5-甲氧基苯并咪唑同时进行缩合和甲氧基化反应制得5-甲氧基-2-[(4-甲氧基-3,5-二甲基吡啶-N-氧化物)-甲硫基]-1H-苯并咪唑. 产物再经三氯化磷还原和高硼酸钠氧化最终制得奥美拉唑,总收率为48.7%. 中间体和奥美拉唑的结构经1H NMR、MS测试技术确证.
【总页数】4页(P178-181)
【作者】万欢;方峰;段梅莉;许煦;冀亚飞
【作者单位】华东理工大学,药学院,上海,200237;华东理工大学,药学院,上
海,200237;华东理工大学,药学院,上海,200237;华东理工大学,化工学院,上
海,200237;华东理工大学,药学院,上海,200237
【正文语种】中文
【中图分类】O626.2
【相关文献】
1.奥美拉唑杂质的合成 [J], 王易;伏世建;刘文杰;张伟
2.有机合成新理念及前沿方法在化学制药工艺学教学中的体现r——以奥美拉唑的合成为例 [J], 高鹏;白梓静
3.奥美拉唑的绿色合成研究 [J], 左慧;马良秀
4.奥美拉唑的合成及拆分 [J], 司玉惠;孙纲春;李志成;李亚萍
5.双氧水催化氧化法合成奥美拉唑的研究 [J], 王宁宁;陈邦
因版权原因,仅展示原文概要,查看原文内容请购买。
×××药业有限公司现行文件奥美拉唑的生产工艺规程文件编号:SOP-MF-301-00起草人:技术员起草日期:年月日审阅人:车间主任审阅日期:年月日审核人:质保经理审核日期:年月日批准人:总经理审批日期:年月日执行日期:年月日分发部门:质量保证部2份生产技术部2份设备部1份目录1、产品概述2、原辅料、包装材料质量标准及规格3、化学反应过程4、生产流程图5、工艺过程6、中间体、半成品的质量标准和检验方法7、技术安全与防火8、综合利用与三废治理9、操作工时与生产周期10、劳动组织与岗位定员11、设备一览表及主要设备生产能力12、原材料、能源消耗定额和技术经济指标13、物料平衡附录附页奥美拉唑的生产工艺规程一:产品概述(一)产品名称1、中文名称:奥美拉唑,别名洛赛克2、英文名称:Omeprazole、Losec3、化学名称:5-甲氧基-2-{[(4-甲氧基-3,5-二甲基-2-吡啶基)-甲基]-亚磺酰基}-1H-苯并咪唑4、分子式:C17H19N3O3S5、分子式量:345.426、化学结构:7、理化性质:本品为白色至类白色结晶性粉末,无臭,遇光易变色,熔点156℃。
本品在二氯甲烷中易溶,在甲醇或乙醇中略溶,在丙酮中微溶,在水中不溶;在0.1mol/L氢氧化钠溶液中溶解。
(二)临床用途①消化性溃疡出血、吻合口溃疡出血。
②应激状态时并发的急性胃黏膜损害,和非甾体类抗炎药引起的急性胃黏膜损伤;③亦常用于预防重症疾病(如脑出血、严重创伤等)胃手术后预防再出血等;④全身麻醉或大手术后以及衰弱昏迷患者防止胃酸反流合并吸入性肺炎。
(三)药理作用本品是近年来研究开发的作用机制不同于H2受体拮抗作用的全新抗消化性溃疡药。
它特异性地作用于胃粘膜壁细胞,降低壁细胞中的氢钾ATP酶的活性,从而抑制基础胃酸和刺激引起的胃酸分泌。
由于氢钾ATP酶又称做"质子泵",故本类药物又称为"质子泵抑制剂"。