溶剂化效应的理论研究计算模型 (1)
- 格式:docx
- 大小:58.31 KB
- 文档页数:12



PCM 溶剂效应原理及计算实例(一)作者:fado 文章来源:本站原创 点击数:669 更新时间:2006-1-28一,处理溶剂效应的理论模型从实验角度而言,溶剂效应主要体现在两个方面,一是溶剂分子与溶质的络合或者氢键等分子间弱相互作用(有时称为“短程”作用),二是极性溶剂通过静电作用对溶质分子电荷分布的影响(“远程”作用)。
对于第一种“短程”作用的理论计算,往往需要运用“真实溶剂模型”(Explicit Solvation Model),即考虑多个溶剂分子与溶质的相互作用,因为这种作用经常由很多不同构型的弱相互作用络合体贡献而得,这种模型的具体应用往往需要结合了蒙特卡罗或分子动力学等方法。
对于第二种“远程”作用,通常需要采用“虚拟溶剂模型”(Implicit Solvation Model)来计算。
现在比较流行的处理方法是引用“反应场”(Reaction Field)的概念,把溶剂效应看成是溶质分子分布在具有均一性质的连续介质(Continuum)当中。
而这种连续介质模型又包含很多分支,PCM (极化连续介质模型,Polari zed Continuum Model)就是最常用的分支之一。
其他一些常用的连续介质模型还包括Onsager, COSMO, SMx(x=1-5)等等。
二,PCM 模型的历史PCM 模型最早由意大利比萨大学的Tomasi 教授于1981年提出,后来又发展出诸多改进版本。
我们先来介绍最初版本的PCM (注:Gaussian 98软件中的关键字SCRF=PCM 即采用该最初版本的PCM )。
三,PCM 溶剂化自由能的构成PCM 模型将溶液中的自由能分为三部分的贡献。
首先,在溶剂分子中建立一个容纳溶质分子的空穴(cavity)会导致体系能量升高。
这部分的能量称为cavity formation energy(比较老的文献中常简称为cavitation energy),这个能量永远是正值(能量升高)。
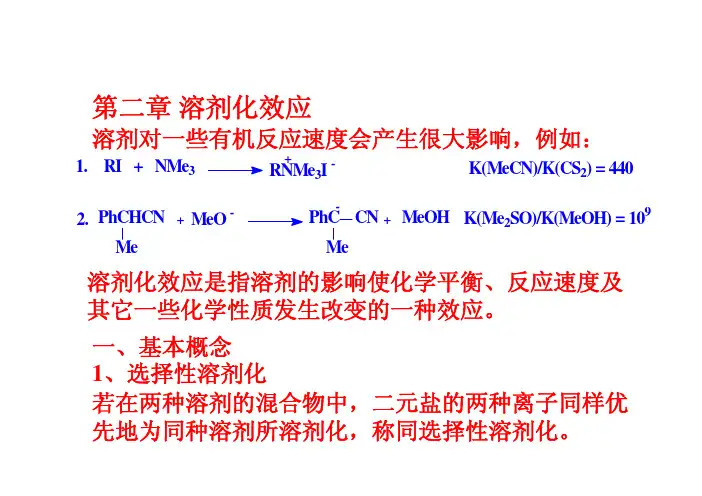
例:CaCl在水-甲醇体系中,Ca2+和Cl–都优先被2水溶剂化。
如阳离子优先被一种溶剂溶剂化,而阴离子优先被另一种溶剂溶剂化,则称异选择性溶剂化。
例:硝酸银在乙腈-水体系中,Ag+优先被乙腈溶剂-优先被水溶剂化。
化, 而NO32、溶剂和溶质分子间的相互作用第一类包括定向诱导力和色散力,这些力是非特异性的,不可能完全饱和。
第二类包括氢键力和电荷转移力,或称电子对授受力。
这类作用有方向并且可以饱和生成化学计量的分子化合物。
C、偶极-诱导偶极力具有永久偶极矩的分子或离子能诱导邻近分子,产生诱导偶极矩,分子在被诱导的瞬间总是处于诱导偶极的方向,两者之间有吸引力。
非极性分子可极化率越大,诱导偶极矩也越大。
这对偶极分子和离子在非极性溶剂中的体系最重要。
D、瞬间偶极-诱导偶极力(色散力〕非极性分子由于电子不断运动,会瞬间产生小的偶极矩,它使邻近分子产生脉冲性极化,从而产生分子间的相互吸引力,这称为色散力。
在两个键已饱和的分子间形成一个附加的成键作用必须是在电子给体分子中存在一个能量足够高的已占据分子轨道,而在电子受体分子中存在一个能量足够底的未占据分子轨道。
3、溶剂的极性和分类(1)质子性溶剂(2)极性非质子性溶剂(3)非极性溶剂量度溶剂极性的标准:(1)偶极矩u常见有机溶剂分子偶极矩的数值在0-5.5D, 在不存在特异性溶质-溶剂间相互作用时,分子极性大小与偶极矩大小一致。
溶剂极性加大,K T 值降低,cis-烯醇式减少。
因为:1、在两种互变异构体中,烯醇式极性较小。
分子内氢键的形成降低羰基偶极-偶极斥力,而在酮式中,这种斥力使其极性加大。
2、分子内氢键使烯醇稳定化,溶剂极性加大,分子间氢键加强,分子内氢键被削弱,烯醇含量减少。
烯醇含量与1,3-二羰基化合物浓度有关。
当偶极性的1,3-二羰基化合物用非极性溶剂稀释,溶剂与羰基作用弱,两羰基偶极斥力大,不稳定;烯醇与溶剂分子间氢键弱,分子内氢键强,烯醇含量增高。


溶剂效应量子化学研究进展摘要:目前计算溶剂效应的方法主要有两类:一种是自洽反应场(SCRF)理论计算方法,该方法从发展初期至今一直应用广泛;另一种是把量子力学(QM)和分子力学(MM)结合起来,分别用量子力学和分子力学来计算化合物和所在溶剂的结构、性质,这种方法近年来发展很快。
本文将通过大量的文献论证了加入溶剂效应所得出的计算结果更加接近真实的实验结果这一结论,以及他们的应用,并提出了溶剂效应理论研究今后的可能发展方向。
关键词:溶剂效应量子化学自洽反应场理论QM/MM模型一、概述化学是研究分子性质和反应的科学,有许多化学过程是在溶液中进行的。
溶剂对分子的构象、电子结构和化学反应有着重要的影响, 因此对溶剂效应进行理论研究有着重要的意义,这更能反映出溶液中分子的真实行为。
所以近年来,越来越多的人开始进入这一领域,并发展了各种不同的理论计算方法。
对这些考虑溶剂效应的方法归结起来有两类,一类是采用经典力场的动力学模拟方法,如:分子动力学(MD)模拟和Monte Carlo模拟。
另一类是采用严格的量子化学计算方法,如:自洽反应场方法(SCRF)、Poisson-Boltzmann方法、AMSOL方法、超分子方法以及量子力学和分子力学相结合的QM/MM方法等。
还有一些方法可以相互结合。
其中自洽反应场理论[1]是目前使用较多的方法,即将静态理论方法和连续介质方法溶剂模型结合起来研究溶液体系。
QM/MM方法[2]是把研究体系设为几个区域,在中心区域进行高精度的量子化学计算(QM),在周围进行半经验或分子动力学计算(MM),这也是处理溶液体系的常用的方法。
本文主要通过围绕这两种方法就近几年来溶剂效应的量子化学计算方法及其应用作一总结。
二、连续介质模型1.自洽反应场理论模型自洽反应场方法(SCRF)是连续介质溶剂模型的一种。
它是目前使用较多的方法,也是本论文重点关注的模型之一,我们将详细介绍。
SCRF方法从Onsager模型发展而来的,孔洞中的偶极分子诱导环绕它的周围介质。




文献综述题目:溶剂化效应的理论研究计算模型作者:xxx班级:xxx学号:xxx日期:2014年5月21号溶剂化效应的理论研究计算模型摘要本文概述了研究溶剂效应计算模型,到目前的溶剂效应的研究进展和研究现状。
介绍了溶剂化效应和描述溶剂效应的一些参数,以及几种重要的溶剂化模型,并分别介绍了基于不同的模型而发展的不同的理论计算方法。
关键词溶剂化效应计算模型综述一、溶剂化现象在溶液中,溶质被溶剂分子包围的现象称为溶剂化,溶剂化作用是溶剂分子通过它们与离子的相互作用,而累积在离子周围的过程。
该过程形成离子与溶剂分子的络合物,并放出大量的热。
溶剂化作用改变了溶剂和离子的结构。
以水溶液为例,其中一个离子周围水的结构模型如图1所示。
图1图中A为化学水化层,该层中由于离子和水偶极子的强大电场作用,使得水分子与离子结合牢固,因而失去平动自由度,这一层水分子和离子一块移动,且水分子数不受温度影响;B为物理水化层,该层水分子也受到离子的吸引,但由于距离较远,吸引较弱,水分子数随温度改变;C为白由水分子层,该层水分子不受离子电场影响。
溶剂不能被单纯宏观地看成一种以密度、介电常数和折射率等物理常数表示特性的连续介质,而应把它看成是单个相互作用的溶剂分子所组成的不连续的介质。
在溶剂及溶液中各质点之间的相互作用,一方面用气体动力学理论的定律处理则太大,而另一方面,如用固体物理的定律来处理则又太小。
溶剂既不是一种使被溶解物在其中扩散以达到紊乱而又均匀分布的惰性介质;也不是象晶体那样具有某种规则结构的介质。
因此气体和晶体两种可能的模型都不能不加限制地应用于溶液。
由于相互作用的复杂性,有关液体的结构,人们所知甚少,甚至最重要的溶剂一水,其内部精细结构的研究也仍然是目前探索的课题frl。
人们提出过许多不同的模型用来描述水的结构,但是所有这些模型均未能圆满地描述水的物理一化学性质和解释水的异常特性。
因此,用实验和理论方法研究液体的结构和相互作用,是物理化学中最为艰巨的任务。
高效液相色谱中溶剂效应理论高效液相色谱是有机分析,特别是药物分析的重要手段。
而色谱峰峰形影响着化合物的定量和定性。
在影响色谱峰峰形的诸多因素中,“溶剂效应”经常被忽视。
本文是对高效液相色谱中“溶剂效应”的漫谈,水平有限,欢迎批评指正。
高效液相色谱已经广泛应用于有机分析,特别是药物分析工作中。
分析工作者通常将更多精力集中在流动相和仪器方法的选择上,忽视了溶解样品所用溶剂的重要性。
高效液相色谱进样之前,必须选择一种合适的溶剂溶解样品。
如果溶剂选择不当则会产生“溶剂效应”,使分析工作者在定性和定量分析中产生误判,影响分析结果。
因此,了解“溶剂效应”产生的原因及掌握避免“溶剂效应”的基本方法对分析工作者是十分必要的。
溶剂效应的概念溶剂效应亦称“溶剂化作用”。
指液相反应中,溶剂的物理和化学性质影响反应平衡和反应速度的效应。
可能造成色谱峰展宽、分叉、保留时间漂移、峰面积变化,双峰等现象。
与此同时,较早洗脱的峰出现前沿或分叉,较晚洗脱的峰峰形正常【1】。
溶剂效应产生的原因样品进入高效液相色谱中,当溶剂与流动相存在差异时,一部分样品溶解进入了流动相,一部分还留在溶剂里,造成色谱保留的差异。
造成这种差异的原因主要有以下几个方面:(1)溶剂强度在反相色谱系统中溶剂强度的顺序为水(最弱)<甲醇<乙腈<乙醇<四氢呋喃<丙醇<二氯甲烷(最强)。
溶解样品的溶剂强度大于该样品出峰时流动相强度,样品溶剂可以看成流动相的一部分,一部分样品溶解于溶剂中会被迅速洗脱出色谱柱,而一部分样品溶解于流动相,被流动相洗脱出,这样会造成色谱峰的展宽或者分叉【2】。
图1是溶剂强度对色谱峰形的影响。
图1 溶剂强度对色谱峰影响[2]图2是笔者采用AgilentZORBAX TC-C18色谱柱以乙腈-水(30:70)为流动相,采用不同的溶剂对目标分析物进行处理制备同一浓度的溶液,考察目标分析物的峰面积及峰高。
图2 不同溶剂溶剂效应对比(2)进样体积进样体积造成的溶剂效应是一般与溶剂种类有关的。
理论计算中溶剂效应模型的构建及应用
戚传松;邓兴旺;李巍
【期刊名称】《北京石油化工学院学报》
【年(卷),期】2006(014)003
【摘要】目前计算溶剂效应的方法主要有两种:一种是自洽反应场(self-consistent reaction field,SCRF)理论计算法,该方法从发展初期至今一直应用广泛;另一类是将量子力学(quantum mechanics,QM)和分子力学(molecular mechanics,MM)结合起来,分别用量子力学和分子力学来计算化合物及所在溶剂的结构与性质,这种方法近几年发展较快.笔者通过大量的文献论证了加入溶剂效应所得出的计算结果更加接近真实的实验结果这一结论.
【总页数】4页(P30-33)
【作者】戚传松;邓兴旺;李巍
【作者单位】北京石油化工学院化学工程系,北京,102617;北京石油化工学院化学工程系,北京,102617;北京石油化工学院化学工程系,北京,102617
【正文语种】中文
【中图分类】O6
【相关文献】
1.应用定标粒子理论计算Menschutkin反应的动力学溶剂效应 [J], 陈六平;韩世钧
2.2-环己烯酮紫外光谱及其溶剂效应的密度泛函理论计算 [J], 李磊;刘鹏;郑银萍;
郑旭明
3.三聚氰胺分子的电子吸收光谱、二阶极化率及其溶剂效应的密度泛函理论计算[J], 周玲;赵改云;张干兵;叶勇;夏清华
4.甲醛和氟代甲醛在不同溶剂中溶剂化效应的理论计算 [J], 刘玲玲;王桂良;张力;常国华;张国富
5.二硝酰胺铵溶剂效应的理论计算 [J], 陈鑫健;胡冬冬;胡一飞;汪营磊;任钟旗因版权原因,仅展示原文概要,查看原文内容请购买。
文献综述题目:溶剂化效应的理论研究计算模型作者:xxx班级:xxx学号:xxx日期:2014年5月21号溶剂化效应的理论研究计算模型摘要本文概述了研究溶剂效应计算模型,到目前的溶剂效应的研究进展和研究现状。
介绍了溶剂化效应和描述溶剂效应的一些参数,以及几种重要的溶剂化模型,并分别介绍了基于不同的模型而发展的不同的理论计算方法。
关键词溶剂化效应计算模型综述一、溶剂化现象在溶液中,溶质被溶剂分子包围的现象称为溶剂化,溶剂化作用是溶剂分子通过它们与离子的相互作用,而累积在离子周围的过程。
该过程形成离子与溶剂分子的络合物,并放出大量的热。
溶剂化作用改变了溶剂和离子的结构。
以水溶液为例,其中一个离子周围水的结构模型如图1所示。
图1图中A为化学水化层,该层中由于离子和水偶极子的强大电场作用,使得水分子与离子结合牢固,因而失去平动自由度,这一层水分子和离子一块移动,且水分子数不受温度影响;B为物理水化层,该层水分子也受到离子的吸引,但由于距离较远,吸引较弱,水分子数随温度改变;C为白由水分子层,该层水分子不受离子电场影响。
溶剂不能被单纯宏观地看成一种以密度、介电常数和折射率等物理常数表示特性的连续介质,而应把它看成是单个相互作用的溶剂分子所组成的不连续的介质。
在溶剂及溶液中各质点之间的相互作用,一方面用气体动力学理论的定律处理则太大,而另一方面,如用固体物理的定律来处理则又太小。
溶剂既不是一种使被溶解物在其中扩散以达到紊乱而又均匀分布的惰性介质;也不是象晶体那样具有某种规则结构的介质。
因此气体和晶体两种可能的模型都不能不加限制地应用于溶液。
由于相互作用的复杂性,有关液体的结构,人们所知甚少,甚至最重要的溶剂一水,其内部精细结构的研究也仍然是目前探索的课题frl。
人们提出过许多不同的模型用来描述水的结构,但是所有这些模型均未能圆满地描述水的物理一化学性质和解释水的异常特性。
因此,用实验和理论方法研究液体的结构和相互作用,是物理化学中最为艰巨的任务。
人们曾设想溶剂和溶质的相互间有种种作用力,由此曾导出数百年的老原理,即“相似相溶”原理。
但是,这一原理是经验的总结,其正确性是有限度的,许多例子中,尽管两种组分结构相似,也可能出现不溶性。
例如,聚乙烯醇不溶于乙醇,醋酸纤维素不溶于乙酸乙酷,聚丙烯睛不溶于丙烯睛等。
另一方面,在结构上不相似的化合物也能形成溶液。
例如,甲醇和苯,水和N、N-二甲基甲酞胺,苯胺和乙醚,聚苯乙烯和三氯甲烷等。
化学是研究分子性质和反应的科学,有许多化学过程是在溶液中进行的。
溶剂对分子的构象、电子结构和化学反应有着重要的影响,因此对溶剂效应进行理论研究有着重要的意义,这更能反映出溶液中分子的真实行为。
例如,气相环境中氨基酸的中性分子结构比两性离子构型更稳定,但在溶液中则是两性离子的构型来得更稳定。
化学,物理学以及生物化学现象的实验反馈在很大程度上依赖于所考虑分子四周的溶剂环境。
因此,人们迫切需要找到一种能计算溶液中分子的电子结构以及性质的方法,并要求这种方法的精度与计算气相分子所能达到的精度相似。
从上世纪七十年代开始,己经出现了许多对分子在溶液中性质的理论方法。
主要是基于两种思想:一种是把注意力集中在有限数目的溶剂分子和溶剂分子的微观相互作用上。
对超分子体系模拟成单个分子来进行计算推断整个体系的性质,即明显处理溶剂一溶质相互作用;第二种基本思想是对溶剂效应引进平均信息,即用具有某种性质,如介电常数、热膨胀系数等的宏观连续介质取代溶剂的微观描述。
二、溶剂化效应的研究现状对于溶剂化效应的研究,最早的最普遍的方法是以统计力学为基础的积分方程理论。
目前,利用积分方程理论,可以对简单液体及少数复杂液体的径向分布函数进行计算,从而求解计算其热力学性质。
但是由于积分方程理论是建立在统计力学的基础之上,所考虑的是所有粒子作用的统计平均效果。
对于这些宏观量的微观机制,人们却知之甚少。
近年来,随着计算机科技的发展,在计算机模拟领域取得了快速的进步,已可用蒙特卡罗(MC)和分子动力学(MD)对简单的单价金属离子的水化进行研究。
以量子化学为基础的计算机模拟,在连续介质模型(SCRF)的基础上发展了许多新的溶剂模型,例如:极化连续介质模型(PCM),积分连续介质模型(IEFPCM),极化导体模型(COSMO,也称CPCM)等。
这些理论模型的程序化,已取得了一些与实验极其吻合的结果,可用来对一些药物分子,蛋白质分子,进行结构优化和稳定性分析,以及一些金属离子的水化和质子转移特性的研究。
并且还出现了多种方法相结合的模拟方法,如:量子力学和蒙特卡洛(MC)方法的结合、量子力学和分子动力学(MD)方法的结合、QM/MM-MD方法、量子力学和光谱学研究的结合。
三、溶剂效应的研究进展自20世纪20年代,Schrodinger, Heisenberg, Dirac等创立的量子力学体系以来,科学家们就开始用量子力学方法处理化学问题。
到了50年代由于计算机的出现,为量子化学提供了有力的工具,分子轨道(MO)理论因易于程序化而蓬勃发展起来。
从70年代开始,MO的从头计算研究逐步展开,80年代,量子化学的研究对象也从中小分子向较大分子、重原子体系发展,随之而来了各种计算方法和程序化,是MO理论和从头算技术大发展时期。
80年代后,以密度泛函为基础的DFT方法也迅速发展起来,它改变了以往以轨道波函数为基的特点,而以密度函数为基。
但直到90年代,随着化学研究对象的不断拓宽,人们不满足于只研究单一的分子或气相中的体系,研究对象发展到固体表面吸附、溶液中的化学反应、生物大分子,单纯的分子轨道方法或密度泛函方法不一定能获得理想结果,这时考虑溶剂效应的量子化学计算方法便发展了起来。
进入21世纪,随着科学研究的深入,化学家己不满足于静态的理论研究,迫切要求计算化学从静态向动态发展,那么分子力学或量子力学与统计力学结合的分子模拟技术填补了这一空白,并获得快速发展,常用于处理溶液体系,成为研究动态过程的有力工具。
对于溶剂效应,按溶剂和溶质相互作用的性质一般可划分成两类,一类是把溶剂看成连续介质,即非特异性溶剂效应。
该模型主要从溶剂对溶质的极化作用角度来考察溶剂的影响,因而也称物理溶剂效应。
另一类是把溶剂看成具体的分子,即特异性溶剂效应。
该模型主要从溶剂和溶质之间的氢键和电荷转移相互作用角度来考察溶剂的影响,因而也称为化学溶剂效应。
基于上述两种模型发展了各种不同的理论方法。
归结起来大致有两类,一类是采用严格的量子化学计算方法,如:自洽反应场方法(Self-Consistent Reaction Field, SCRF), Poisson-Boltzmann方法、AMSOL方法、超分子方法(Super molecule)以及量子力学与分子力学相结合的QM/MM方法等。
另一类是采用经典力场的动力学模拟方法,如分子动力学(Molecular Dynamic, MD)模拟和Monte Carlo模拟等。
以上诸方法各有特色和优缺点,有些方法还可以相互结合。
其中自洽反应场方法是目前使用较多的考虑溶剂效应的方法,即将静态理论方法和连续介质溶剂模型结合起来研究溶液体系。
Poisson-Boltzmann方法就是利用求解PB方程来计算溶质与溶剂分子间的静电极化作用。
AMSOL方法采用Born模型,进行分子轨道方法计算,来研究溶液体系。
超分子方法认为溶剂分子可能通过氢键或电荷转移作用与溶质分子紧密结合在一起,然后将这些“额外”原子包含在一个更大的但仍是孤立的体系中进行处理。
QM/MM方法fuel是把研究体系设为几个区域,在中心区域进行高精度的量子化学计算(QM),在周边区域进行半经验或分子力学计算(MM),该方法也是处理溶液体系的一个较好的方法。
分子动力学或Monte Carlo模拟溶液体系,最适用于研究溶质分子周围溶剂分子的结构性质,而对溶剂化自由能的计算则没有自洽反应场方法计算得到的结果精确。
四、溶剂化理论研究计算模型从上世纪七十年代开始,已经出现了许多对分子在溶液中性质的理论方法。
主要是基于两种思想:一种是把注意力集中在有限数目的溶剂分子和溶剂分子的微观相互作用上。
对超分子体系模拟成单个分子来进行计算推断整个体系的性质,即明显处理溶剂-溶质相互作用;第二种基本思想是对溶剂效应引进平均信息,即用具有某种性质,如介电常数、热膨胀系数等的宏观连续介质取代溶剂的微观描述。
计算量子化学中描述溶剂对溶质极化作用的方法有:超分子方法,量子力学-分子力学联合方法(QM-MM),自洽反应场方法(SCRF)。
其它的溶剂化方法还有:基于统计力学的参考反应区模型(RISM),有效碎片势(EFP)和ONIOM-XS方法。
1、分子簇模型直接溶剂化模型把注意力集中在有限数目的溶剂分子和溶剂分子的微观相互作用上。
用分子簇模型这种方法来研究在分子水平上的溶质分子间的相互作用、溶质和溶剂分子间的相互作用。
利用这种方法Dong-Mei Huang和Yi-Bo Wang研究了吠喃和卤化氢的相互作用;陶福明等研究了分子在水中的离解反应,通过逐个加水分子的方法研究水对分子电离的溶剂化效应。
如:溴化氢、氟化氢、等都可以用此方法来研究水对分子电离的溶剂化效应,并被证实此方法是可靠的。
应用表明,此方法除了可以用于无机分子的研究,还可以用于有机分子的研究,如考察2-氯苯酚分别与水分子和氨分子作用的酸度电离平衡常数PKa。
但由于计算量耗费的限制,体系中只能简单的包含少数溶剂分子,因此不适用于考虑大量溶剂分子的影响。
2、自洽反应场方法(SCRF)自洽反应场方法(SCRF)就是对溶剂效应引进平均信息,考虑大量溶剂分子的影响。
为了研究溶剂效应,Onsager提出了反应场理论(Reaction Field Theory)。
在这个模型中,溶质被放在一个沉浸在介电常数为。
的连续介质的空穴中,这个空穴可以是球形、椭圆形或者其它形状,为计算方便起见,通常选用球形。
Tapia和Goscinski将Onsager反应场模型加入量子化学分子轨道理论中,发展成为自洽反应场(SCRF)理论。
此后,这一模型被广泛地应用于溶剂效应的研究中。
SCRF方法将溶剂分子看作连续介质,溶质分子使介质发生极化并产生一个反应场,后者又反过来使溶质分子本身发生极化。
采用SCRF方法,只需适当选取介电常数就可以描述分子在不同溶剂中的行为,从而研究分子在溶剂中的水化性质。
与直接溶剂模型相比,连续介质模型至少有两个优势。
首先,它能够显著地减少所研究的体系的自由度。
第二个优势是其能够方便准确地处理最重要的长程静电作用的体系,这种作用是许多溶剂化现象的主要促成因素。
因此,基于连续介质模型的SCRF理论现在己经被广泛地用于研究溶剂对溶质分子结构和性质的影响以及溶液中的化学反应。