系统进化树的制作
- 格式:ppt
- 大小:2.37 MB
- 文档页数:36
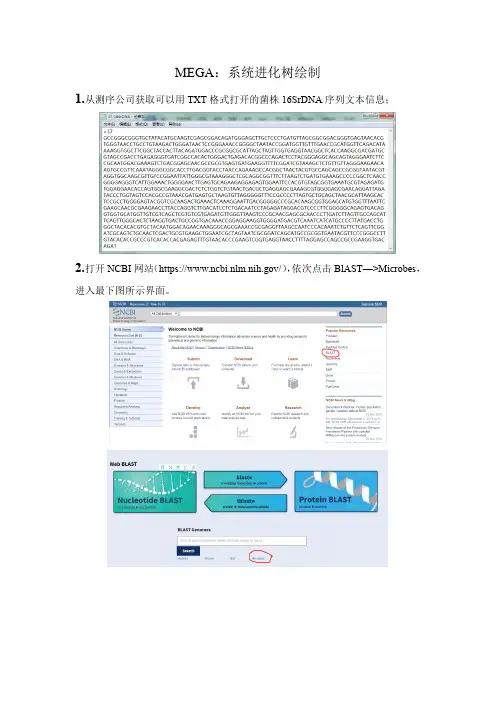
MEGA:系统进化树绘制1.从测序公司获取可以用TXT格式打开的菌株16SrDNA序列文本信息;2.打开NCBI网站(https:///),依次点击BlAST—>Microbes,进入最下图所示界面。
3.将序列信息粘贴到黄色文本框内,点击BLAST按钮,进入比对结果页面,根据所需选择20条(参考)相似序列信息,点击Download,下载FASTA(aligned sequences)格式序列信息到电脑;4.将测序所得序列信息与Blast所得序列信息合并到同一个Text文件中;5.打开MEGA软件(以MEGA6.06为例),点击Align—>Edit/Build Alignment,选择Creat a new alignment并点击OK,点击DNA,进入最下图界面,最大化子界面;6.点击下图红线圈出的图标或通过Edit—>Insert Sequence From File Ctrl+I,进入第二个图所示界面,将文件格式由ABI改为Text,选择所选序列信息文件,点击打开;7.按住Shift,鼠标点击首条和最后一条多余的序列信息,即可选择某一需要删除的序列信息区域,点击Delete删除多余序列信息,并编辑各序列名称,点击保存编辑好的序列信息;8.点击Data—>Phylogenetic Analysis(系统进化分析),点击“Yes”完成系统进化分析;9.回到MEGA主页面,依次点击Analysis—>Phylogeny—>Construct/Test Neighbor-Joining Tree...,在跳出的界面中点击“Yes”,接着将跳出页面中的Test of Phylogeny项的None改为Bootstrap method,点击Compute,系统完成运算,生成系统进化树;10.依次点击Image—>Save as PDF file,保存成PDF格式系统发育树图谱。

系统进化树的构建方法系统进化树(systematic phylogenetic tree)是用于描述不同物种之间进化关系的一种图形化表示方法,可以帮助我们理解物种的起源、演化和分类。
构建系统进化树主要涉及到物种的分类学和进化生物学知识,以及系统发育分析方法。
下面将介绍系统进化树的构建方法。
1.选择研究对象:确定研究的物种范围,通常会选择有代表性的物种,包括已知的和新发现的物种。
2.收集DNA序列数据:从每个研究对象中提取DNA样本,并通过PCR扩增得到所需的基因序列。
常用的基因包括线粒体基因COI、核基因ITS 等,根据具体研究目的和对象进行选择。
3.序列比对:将收集到的DNA序列进行比对,通常采用计算机程序进行全局比对,比对结果会显示序列之间的同源区域和差异。
4. 构建系统进化树:有多种方法可以构建系统进化树,其中最常用的是系统发育建模方法,如最大简约法(maximum parsimony)、最大似然法(maximum likelihood)和贝叶斯推断(Bayesian inference)等。
最大简约法是最简单和最常用的构建系统进化树的方法之一、它基于简约原则,认为进化过程中最少的演化步骤是最可能的。
方法将不同物种的序列进行比对,统计共有的字符以及不同的字符,根据最小化改变的原则,得到进化树。
最大似然法使用概率模型来计算物种之间的进化关系,根据序列数据的概率分布确定最可能的进化树。
这种方法考虑了不同序列字符的不同演化速率以及序列之间的相关性。
贝叶斯推断方法基于贝叶斯统计学原理,通过计算不同进化树的后验概率来确定最有可能的进化树。
该方法能够对不同进化模型和参数进行全面的推断,但计算复杂度较高。
5.进行分支长度调整和进化树根的定位:进化树的分支长度表示物种间的差异,可以根据各个物种间的差异大小进行调整。
进化树的根通常是已知的进化历史或已知的进化事件,如灭绝事件等,可以通过分析群体间的基因流动等信息进行推断。

1.MEGA构建系统进化树的步骤2.CLUSTALX进行序列比对1.MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
如图:2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图:。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图:。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes,如图:。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,如图:6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。

作系统进化树的方法系统进化树(Phylogenetic tree)是一种表示生物物种之间进化关系的图形结构。
它基于生物的遗传物质或形态特征等数据,通过一定的算法和模型来构建,以揭示物种之间的亲缘关系和进化历程。
以下是构建系统进化树的一般步骤:1. 数据收集:首先需要收集用于构建进化树的基因或形态特征数据。
这通常涉及从各种来源获取DNA、蛋白质或其他分子序列数据,或者从博物馆和标本馆获取生物形态特征数据。
2. 序列比对:对于DNA或蛋白质序列数据,需要将这些序列进行比对,以确保它们可以一起进行比较和分析。
3. 选择适当的距离度量:在构建系统进化树时,需要计算物种之间的“距离”。
这些距离是基于序列或形态特征的差异来计算的。
有多种方法可以计算这些距离,例如基于遗传物质的p距离(代表两个序列之间的差异比例)或形态特征的欧几里得距离。
4. 选择合适的建树算法:系统进化树可以通过多种算法来构建,包括但不限于UPGMA(Unweighted Pair Group Method with Arithmetic Mean)、WPGMA(Weighted Pair Group Method with Arithmetic Mean)、WPGMC(Weighted Pair Group Method with Centroid Linkage)、Neighbor Joining、Fitch-Margoliash、Maximum Parsimony、Maximum Likelihood等。
选择哪种算法取决于你的具体需求和所处理数据的性质。
5. 构建系统进化树:使用选择的算法和距离度量,将物种按照它们的亲缘关系分组。
这一步通常涉及到一个迭代过程,其中算法会尝试不同的分组方案,直到找到一个最优解。
6. 评估和验证树:一旦构建了系统进化树,就需要对其进行评估和验证,以确保其合理性和可靠性。
这通常涉及使用多种统计测试和可视化工具,例如Bootstrapping、P-distance、Tree-bisection-reconnection (TBR) 操作等。



系统进化树的构建一、什么是系统进化树系统进化树,又称为生命进化树或物种树,是描述生物进化关系的一种图形表达方式。
它通过比较不同物种之间的形态、生理特征以及遗传信息等多方面的数据,将它们按照演化顺序排列在一个分枝结构图中,以展示各个物种之间的亲缘关系和演化历程。
二、系统进化树的构建方法1. 形态学比较法形态学比较法是最早被使用的构建系统进化树的方法。
该方法主要通过对不同物种之间形态特征的比较,确定它们之间的亲缘关系。
例如,通过对鸟类翅膀长度和颜色等特征进行比较,可以确定它们之间的亲缘关系,并将它们排列在一个分枝结构图中。
2. 分子生物学方法随着分子生物学技术的发展,越来越多的研究者开始使用DNA序列等遗传信息来构建系统进化树。
这种方法主要是通过比较不同物种DNA 序列或蛋白质序列之间的差异性,来推断它们之间的亲缘关系。
例如,通过对人类、猩猩和大猩猩的DNA序列进行比较,可以确定它们在进化过程中的亲缘关系。
3. 综合方法综合方法是将形态学比较法和分子生物学方法结合起来,以获得更准确的系统进化树。
该方法主要是通过对不同物种之间形态特征和遗传信息等多方面的数据进行综合分析,来推断它们之间的亲缘关系。
例如,通过对恐龙化石的形态特征和DNA序列进行比较,可以确定它们在进化过程中的亲缘关系。
三、系统进化树的构建步骤1. 收集数据构建系统进化树需要收集大量的数据,包括形态特征、遗传信息等多方面的数据。
这些数据可以通过实验、文献调查等方式获取。
2. 数据处理收集到的数据需要进行处理和分析,以便于构建系统进化树。
这些处理包括序列比对、计算差异性等操作。
3. 构建树型结构在经过数据处理后,就可以开始构建系统进化树了。
该步骤主要是将不同物种之间的亲缘关系按照演化顺序排列在一个分枝结构图中。
4. 树型验证构建完系统进化树后,需要对其进行验证。
这可以通过计算分支长度、计算拓扑稳定性等方式来实现。
四、系统进化树的应用1. 生物分类学研究系统进化树可以帮助生物学家更准确地确定不同物种之间的亲缘关系,从而更好地进行生物分类学研究。

邻接法构建系统进化树
邻接法是一种常见的构建系统进化树的方法。
该方法基于分子序列分析,通过比较序列之间的相似性来推断它们的进化关系。
具体来说,邻接法根据序列间的距离矩阵,将序列之间的关系表示为一个无向图。
然后,该方法根据图的拓扑结构,以最小化进化的总分支长度为目标,逐步合并节点,构建系统进化树。
与其他构建系统进化树的方法相比,邻接法具有计算简便、运算速度快等优点,因此被广泛应用于生物信息学领域。
但是,邻接法也存在一些缺点,如容易受到序列长度和序列间的不均衡分布的影响,导致结果不够准确。
因此,在应用邻接法构建系统进化树时,需要谨慎选择序列和进行适当的处理,以获得更可靠的结果。
- 1 -。

MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA 序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。

邻接法构建系统进化树系统进化树是遗传学和分类学中重要的概念之一,它反映了生物物种在进化历程中的演化关系和分类关系。
构建系统进化树是一项复杂而具有挑战性的任务,需要研究者通过多种途径收集并整理大量的生物学数据,然后运用适当的方法对这些数据进行分析和整合,最终得出一个可信的系统进化树。
邻接法是构建系统进化树的一种常用方法,它是基于生物物种之间相似性程度来构建树形结构的。
邻接法通过计算生物物种之间的相似性指标来确定物种之间的亲缘关系。
这些相似性指标可以是形态学、生物化学、分子生物学等多种生物学特征。
由于这些相似性指标具有不同的权重和精度,所以在邻接法中需要对它们进行合理的加权和处理。
在邻接法中,首先需要构建一个物种之间的相似性矩阵。
这个矩阵是一个方形矩阵,其中每一行和每一列分别代表不同的生物物种,矩阵的元素是两个物种之间的相似度指标。
在构建这个矩阵时,需要用适当的算法计算不同生物特征之间的相似性,然后将它们组合成一个综合的相似性指标。
一旦建立了物种之间的相似性矩阵,邻接法就可以应用了。
在邻接法中,首先需要将物种两两配对,然后分别计算相似度矩阵中它们之间的相似度指标,并将它们连接起来形成初步的树形结构。
接下来,邻接法会找到相似度矩阵中最高的相似度指标,将对应的物种节点连接起来。
然后,需要重新计算这些已连接的节点与其他未连接节点之间的相似度指标。
这个过程会一直持续到所有的物种节点都被连接起来,最终形成一棵完整的系统进化树。
邻接法构建系统进化树的优点在于它简单易行,能够快速生成一个初步的进化树,常常被人们用于构建大规模的分类系统。
但邻接法也存在一些局限性,例如在处理复杂的进化关系时会产生误差,而且它无法反映物种之间细微的差异和不同方面的进化过程。
因此,在应用邻接法构建系统进化树时需要对具体的应用场景和数据特征进行充分的了解和评估,来保证得到可靠和准确的系统进化树。
系统发育进化树构建系统发育进化树(Phylogenetic tree)是一种用于描述物种或群体之间进化关系的图形表示。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
本文将介绍系统发育进化树的构建方法和其在生物学领域中的应用。
一、系统发育进化树的构建方法1. 选择合适的基因或序列:构建系统发育进化树需要选择适当的基因或序列进行分析。
常用的基因包括核糖体RNA(rRNA)和线粒体DNA(mtDNA)等。
2. 收集物种样本:从不同物种中收集样本,并提取相应的基因或序列。
3. 序列比对:将收集到的序列进行比对,找出它们之间的相同和差异。
4. 构建进化模型:根据序列比对的结果,选择适当的进化模型,如最大似然法或贝叶斯推断等。
5. 构建进化树:利用选定的进化模型,根据序列的相似性和差异性,构建系统发育进化树。
二、系统发育进化树的应用1. 物种分类:系统发育进化树可用于物种分类,帮助我们理解不同物种之间的亲缘关系。
通过比较进化树上的分支长度和节点位置,我们可以判断物种之间的相似性和差异性。
2. 进化研究:系统发育进化树可用于研究物种的进化历史和进化速率。
通过比较不同物种之间的进化树,我们可以了解它们的共同祖先以及它们之间的演化路径。
3. 分子演化研究:系统发育进化树在分子演化研究中起着重要的作用。
通过比较不同物种的基因或序列,我们可以推断它们的演化历史和演化速率。
4. 物种保护:系统发育进化树可用于指导物种保护工作。
通过研究物种的进化关系,我们可以了解哪些物种是濒危物种或有特殊保护需求的物种。
5. 药物开发:系统发育进化树可用于药物开发。
通过比较不同物种的基因或序列,我们可以了解它们之间的差异,并找到可能具有药用潜力的物种。
总结:系统发育进化树是一种重要的工具,用于描述物种或群体之间的进化关系。
通过构建系统发育进化树,我们可以了解不同物种之间的亲缘关系,以及它们的共同祖先。
系统发育进化树在物种分类、进化研究、分子演化研究、物种保护和药物开发等领域都有着广泛的应用。
系统进化树的构建1. 引言在计算机科学领域,系统进化树是一种用于描述和分析软件系统演化历史的工具。
它可以帮助我们理解软件系统是如何随着时间发展和演变的,以及不同版本之间的关系。
通过构建系统进化树,我们可以更好地了解软件系统的演化规律,为软件维护、升级和迭代提供有效的指导。
本文将详细介绍系统进化树的构建方法,并提供相关示例和实践经验。
2. 构建方法2.1 数据收集构建系统进化树的第一步是收集相关数据。
这些数据可以来自于版本控制系统、缺陷跟踪系统、代码仓库等多个来源。
主要包括以下几个方面:•版本信息:记录每个版本的发布日期、版本号等基本信息。
•变更集:记录每个版本中进行了哪些变更,包括新增功能、修改bug等。
•缺陷报告:记录每个版本中出现的缺陷报告,包括缺陷编号、严重程度等。
•代码仓库:记录每个版本中所使用的代码库快照。
2.2 数据预处理在进行数据分析之前,需要对收集到的数据进行预处理。
主要包括以下几个方面:•数据清洗:去除重复、无效或不完整的数据。
•数据整合:将不同来源的数据进行整合,建立关联关系。
•数据格式化:将数据转换为统一的格式,方便后续分析和处理。
2.3 构建演化关系构建系统进化树的核心是建立不同版本之间的演化关系。
可以使用以下两种方法来实现:2.3.1 基于变更集通过分析每个版本中的变更集,可以识别出新增、修改和删除的功能模块或代码文件。
根据这些变更信息,可以构建出一个版本间的差异图,从而揭示出系统演化的路径。
2.3.2 基于缺陷报告通过分析每个版本中出现的缺陷报告,可以识别出哪些缺陷被修复,并确定修复缺陷所涉及到的代码文件或功能模块。
根据这些信息,可以构建出一个修复路径图,从而揭示系统演化过程中缺陷修复的路径。
2.4 可视化展示构建完成系统进化树后,需要将其以可视化形式展示出来。
常用的可视化工具有网络图、树状图等。
通过可视化展示,可以更直观地了解系统的演化历史和各个版本之间的关系。
3. 示例与实践经验3.1 示例以一个开源软件项目为例,假设我们收集到了该项目的版本控制记录、缺陷报告和代码仓库快照。
邻接法构建系统进化树
邻接法是一种常用的构建系统进化树的方法。
该方法通过研究不同物种之间的形态、生理、生态等方面的差异,以及它们的遗传距离和进化历史,将它们之间的关系以树形结构表示出来,以更好地理解和研究它们之间的演化关系。
邻接法的核心思想是基于“邻接矩阵”的概念来构建进化树。
邻接矩阵是一种方阵,其中每个元素代表两个物种之间的相似性或差异性。
通过计算不同物种之间的相似性或差异性,可以得到一个N×N 的邻接矩阵,其中N代表物种数目。
在构建进化树的过程中,邻接矩阵会不断被更新和改变。
首先,根据邻接矩阵中不同物种之间的相似性或差异性,可以将相似性较高的物种聚为一类。
然后,通过计算不同类之间的相似性或差异性,可以得到新的邻接矩阵。
这个过程不断迭代,直到只剩下一个类为止。
最终,得到的进化树就是基于邻接矩阵构建出来的。
邻接法构建系统进化树的优点是计算速度快,而且结果可靠。
与此同时,它也存在一些缺陷,比如无法处理物种之间的多样性,以及缺乏模型支持等问题。
因此,在使用邻接法时,需要根据研究的具体问题和数据特点来选择合适的方法。
- 1 -。