遗传修饰基因与地中海贫血表型
- 格式:pptx
- 大小:18.67 MB
- 文档页数:4

遗传修饰与表观遗传遗传修饰和表观遗传是生物学中重要而又复杂的概念。
它们在个体的发育过程中起着至关重要的作用。
本文将详细介绍遗传修饰和表观遗传的概念、机制以及它们对生物多样性的影响。
一、遗传修饰的概念和机制遗传修饰是指个体的基因组在发育过程中发生的可逆性、可遗传性地改变。
它能够通过调控基因的表达来适应环境变化和维持生命活动的平衡。
遗传修饰主要包括DNA甲基化和染色质修饰两种形式。
1. DNA甲基化DNA甲基化是指DNA分子上的甲基基团与碱基胞嘧啶结合的化学修饰过程。
它主要发生在CpG位点上,通过酶的催化作用实现。
DNA 甲基化在调控基因的表达和稳定基因组结构方面起着重要作用。
甲基化的程度和位置可以影响DNA的结构和功能,从而影响基因的表达模式。
2. 染色质修饰染色质修饰是指染色质分子上的化学修饰,包括乙酰化、甲基化、磷酸化等。
这些修饰可以影响染色质的结构和紧密度,从而对基因的表达进行调控。
染色质修饰主要通过核小体组装、染色质层次结构调整以及转录因子的结合来实现。
二、表观遗传的概念和机制表观遗传是指不涉及DNA序列改变的遗传信息传递。
它通过遗传修饰等机制影响基因的表达,进而影响个体的性状和适应性。
表观遗传主要包括DNA甲基化、组蛋白修饰和非编码RNA等。
1. DNA甲基化与表观遗传DNA甲基化作为一种常见的表观遗传修饰形式,可以通过影响基因的甲基化模式来调控基因的表达。
在细胞分化过程中,DNA甲基化可以编码细胞的记忆,继而影响细胞分化和功能特化。
2. 组蛋白修饰与表观遗传组蛋白修饰是指对组蛋白分子进行化学修饰,从而影响染色质结构和基因的表达。
组蛋白修饰主要通过改变染色质状态来调控基因转录的进行,进而影响个体发育和适应环境。
3. 非编码RNA与表观遗传非编码RNA是指不参与蛋白质合成的RNA分子,包括长链非编码RNA(lncRNA)、微小RNA(miRNA)等。
这些非编码RNA可以通过干扰RNA的转录和翻译过程,调控基因的表达水平和模式。
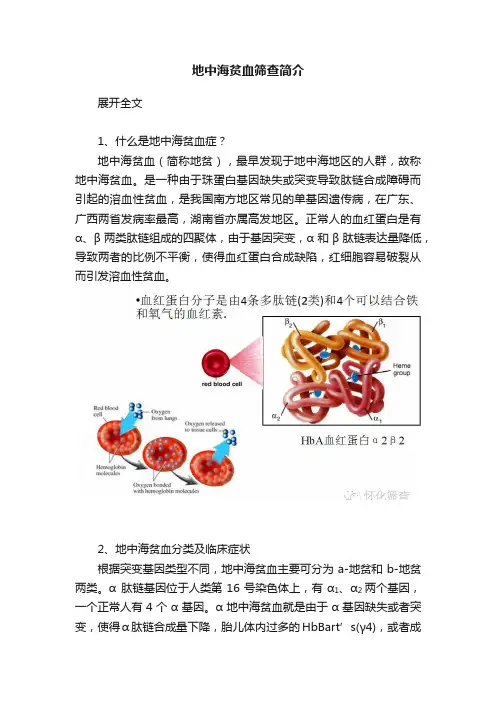
地中海贫血筛查简介展开全文1、什么是地中海贫血症?地中海贫血(简称地贫),最早发现于地中海地区的人群,故称地中海贫血。
是一种由于珠蛋白基因缺失或突变导致肽链合成障碍而引起的溶血性贫血,是我国南方地区常见的单基因遗传病,在广东、广西两省发病率最高,湖南省亦属高发地区。
正常人的血红蛋白是有α、β两类肽链组成的四聚体,由于基因突变,α和β肽链表达量降低,导致两者的比例不平衡,使得血红蛋白合成缺陷,红细胞容易破裂从而引发溶血性贫血。
2、地中海贫血分类及临床症状根据突变基因类型不同,地中海贫血主要可分为a-地贫和b-地贫两类。
α 肽链基因位于人类第16号染色体上,有α1、α2两个基因,一个正常人有4个α基因。
α地中海贫血就是由于α基因缺失或者突变,使得α肽链合成量下降,胎儿体内过多的HbBart’s(γ4),或者成人体内过多的β珠肽链形成HbH(β4),导致无效造血和红细胞破坏。
引发α地中海贫血的突变在我国主要有三种缺失和三种突变。
分别是缺失型——SEA/;-α3.7/;-α4.2/及点突变αCS;αWS,αQS。
根据α基因的缺失情况及临床症状α地中海贫血又可分为静止型、轻型、中间型和重型。
一般来说只缺失一个α基因,成人基本无明显临床表现,部分携带者血常规和血红蛋白电泳无异常。
而对于缺失两个α基因的携带者,会有轻微的贫血,其血常规和血红蛋白电泳都有一些异常,这样的携带者如果和同样缺失α基因的患者结婚,就有可能会生育重型地贫患儿。
轻型携带者与静止性携带者有四分之一的可能生育HbH患儿,就是缺失3个α基因,是中间型地贫患者,有较为明显的贫血,临床表型因人而异,有的HbH患者能够比较好的生活,而有的则有比较严重的贫血,影响到生活质量。
轻型α地贫患者若与同样是轻型α地贫患者结婚则有四分之一的可能生育重型地贫患儿即HbBart’s患儿。
HbBart’s 患儿即缺失四个α基因,无法存活。
HbBart’s 胎儿会产生严重的水肿胎现象,受累胎儿由于严重贫血、缺氧常于妊娠23-40周左右在宫内或分娩半小时后死亡。


地中海贫血基因遗传规律地中海贫血是一种常见的遗传性疾病,主要发生在地中海、中东、北非等地区。
该疾病是由于基因突变导致体内血红蛋白合成受到影响,引起贫血等症状。
本文将介绍地中海贫血的基因遗传规律,包括遗传方式、突变类型和基因检测等方面。
一、遗传方式地中海贫血是一种常染色体隐性遗传疾病。
人类体细胞中有23对染色体,其中有一对性染色体。
男性有XY染色体,女性有XX染色体。
地中海贫血基因位于第11号染色体上,是由HBB基因编码的血红蛋白β链所致。
一个人有两个HBB基因,一个来自父亲,一个来自母亲。
如果两个基因都正常,那么这个人就不会得地中海贫血。
如果一个基因正常,一个基因携带突变,那么这个人是携带者,不会得地中海贫血,但有可能将突变基因传给后代。
如果两个基因都携带突变,那么这个人就会得地中海贫血。
二、突变类型地中海贫血是由多种不同类型的突变引起的。
最常见的突变类型是点突变,即基因序列中的一个碱基发生突变。
点突变包括错义突变、无义突变和同义突变。
错义突变是指突变后的基因编码的氨基酸与原来不同,导致血红蛋白的结构和功能发生改变,从而引起地中海贫血。
无义突变是指突变后的基因编码的氨基酸变为终止密码子,导致基因转录过程提前终止,从而缺失血红蛋白β链的一部分,引起地中海贫血。
同义突变是指突变后的基因编码的氨基酸与原来相同,但可能会影响基因转录和翻译,从而导致地中海贫血。
除了点突变,地中海贫血还可能由缺失、插入和重复等突变引起。
缺失突变是指基因序列中丢失了一段碱基,导致血红蛋白β链缺失一部分,引起地中海贫血。
插入突变是指基因序列中插入了一段碱基,导致血红蛋白β链多出一部分,引起地中海贫血。
重复突变是指基因序列中某些碱基重复出现,导致血红蛋白β链中某些氨基酸重复出现,引起地中海贫血。
三、基因检测地中海贫血是一种遗传性疾病,可以通过基因检测来诊断和筛查。
基因检测可以检测HBB基因的突变情况,从而确定一个人是否患有地中海贫血或是携带突变基因。

HKαα地中海贫血的基因诊断及临床表型分析杜丽 王继成 秦丹卿 胡听听 袁腾龙 张艳霞 梁驹卿 尹爱华(广东省妇幼保健院医学遗传中心、广东省妇幼代谢与遗传病重点实验室,广东广州511442)【摘要】 目的 对HKαα地中海贫血病例进行基因诊断,分析HKαα病例临床表型,为遗传咨询提供指导。
方法 对在本院就诊的HKαα病例进行血常规、血红蛋白电泳、基因诊断及产前诊断,分析病例的临床表型。
结果 共检测出HKαα病例33例,包括HKαα/αα15例、--SEA/HKαα16例、αWSα/HKαα1例、-α4.2/HKαα1例。
并对5个家庭进行了产前诊断,检出--SEA/HKαα胎儿1例,通过遗传咨询,避免了选择性终止妊娠。
结论 结论HKαα地中海贫血的基因诊断可以减少病例的漏诊或误诊,临床表型的分析可以为精准的遗传咨询提供指导。
【关键词】 地中海贫血;基因诊断;HKαα【中图分类号】 R714.55 【文献标识码】 A犇犗犐:10.13470/j.cnki.cjpd.2017.03.004 通讯作者:尹爱华,E mail:yinaiwa@vip.126.com【犃犫狊狋狉犪犮狋】 犗犫犼犲犮狋犻狏犲 ToinvestigatethegenediagnosisandclinicalpresentationofHKααthalassemiaandtoprovideaneffectivegeneticcounseling.犕犲狋犺狅犱 Wholebloodcellanalysis,capillaryzoneelectro phoresis(CZE),Gap PCRandpolymerasechainreaction reversedotblot(PCR RDB)assaywereper formedtotheHKααThalassemia,andanalyzethehematologicalphenotypedataofHKααThalassemia.犚犲狊狌犾狋狊 33casesofHKααthalassemiaweredetected,including15casesofHKαα/αα、16casesof--SEA/HKαα、1caseofαWSα/HKααand1caseof-α4.2/HKαα.5pedigreeswerecarriedoutprenataldiagnosisand1fetusofHb--SEA/HKααwasdetected.Thecouplechoosedtocontinuethepregnancy.犆狅狀犮犾狌狊犻狅狀狊 GenediagnosisofHKααthalassemiacanmakeaccuratediagnosis,avoidingthemisdiagnosis.Theanaly sisofHKααthalassemiaclinicalpresentationcanprovideaneffectivegeneticcounseling.【犓犲狔狑狅狉犱狊】 thalassemia;genediagnosis;HKαα α地中海贫血(简称α地贫)是我国南方各省携带率最高、影响最大的遗传病,广东省育龄人群α地贫的基因携带率达13%[1]。

引起地中海贫血症基因功能异常的分子机理概述及解释说明1. 引言1.1 概述地中海贫血症是一种常见的遗传性血液疾病,主要发生在地中海沿岸地区以及亚洲、非洲等部分地区。
它是由于人体红细胞中的血红蛋白异常导致的一系列临床症状和贫血表现。
1.2 文章结构本文将从几个层面上探讨引起地中海贫血症基因功能异常的分子机理,主要包括贫血症及地中海贫血症介绍、分子机理与遗传变异的关系、分子机理的探索与解释以及最后总结与展望。
1.3 目的本文旨在通过对引起地中海贫血症基因功能异常的分子机理进行概述和解释,增进我们对这一遗传性疾病产生原因和发展过程的认识。
同时,希望为未来相关研究提供有关方法和方向上的启示,并推动更多人对该领域进行深入探索。
以上是根据题目所给文章目录初步撰写的“引言”部分内容,请根据需要进行修改和补充。
2. 贫血症及地中海贫血症介绍:2.1 贫血症概述:贫血症是一种常见的血液疾病,其主要特征是体内红细胞数量或功能不足,导致氧输送能力下降。
贫血症可由多种原因引起,包括遗传因素、营养缺乏、慢性疾病、药物副作用等。
这种疾病严重影响着患者的生活质量和健康,尤其在儿童和孕妇中更为常见。
2.2 地中海贫血症简介:地中海贫血症是一种常见的遗传性贫血症,在地中海沿岸国家特别高发。
它由人类红细胞β-珠蛋白基因的变异引起,导致红细胞生成受阻和异常溶解。
地中海贫血症根据临床表型的差异分为两种主要类型:地中海贫血(β-珠蛋白链缺失型)和地中海型α-珠蛋白缺失型。
其中,地中海贫血是最常见的一种。
2.3 地中海贫血症的流行与影响:地中海贫血症主要分布在地中海沿岸地区,特别是意大利、希腊、土耳其和北非等国家。
由于该疾病的遗传性质,携带异常基因的人口比例较高。
这导致了在这些地区有相当多的患者和携带者存在。
地中海贫血症给患者的生活造成了极大困扰,严重影响其生活质量和工作能力。
由于缺乏足够健康的红细胞,身体不断经历衰竭和氧供应不足。
常见的症状包括持续性乏力、呼吸困难、黄疸以及骨骼畸形等。

基金项目:深圳市医疗卫生三名工程(SZSM201601062);深圳市医学重点学科建设资助项目(bZXK054)作者简介:李育敏,女,1982年生,硕士,主治医师,主要从事血红蛋白相关疾病研究。
通信作者:张秀明,E-mail :。
3种β缺失型地中海贫血基因型与表型分析李育敏1, 蔡钦泉1, 覃俊龙1, 金 潇2, 陈亚琼1, 莫云均1, 李 悦1, 张秀明1(1.深圳市罗湖区人民医院医学检验科,广东 深圳 518001; 2.深圳市罗湖区人民医院感染管理科,广东 深圳 518001)摘要:目的 分析中国型G γ+(A γδβ)0、东南亚型-遗传性持续性胎儿血红蛋白血症(HPFH )和台湾型3种β缺失型地中海贫血(简称地贫)的基因型与表型,为地贫筛查提供参考。
方法 选取行血红蛋白(Hb )电泳检测的患者72 397例,采用跨越断裂点-聚合酶链反应和聚合酶链反应-反向斑点杂交法对HbF ≥5%、年龄≥6个月的患者进行常见地贫基因和3种罕见β缺失型地贫分型检测及红细胞参数检测、DNA 测序。
结果 检出19例中国型G γ+(A γδβ)0、18例东南亚型-HPFH 和3例台湾型地贫患者。
单纯中国型G γ+(A γδβ)0 杂合子地贫患者的红细胞平均体积(MCV )、平均红细胞血红蛋白含量(MCH )、HbF 和HbA 2水平分别为(69.1±2.2)fL 、(22.7±1.1)pg 、(15.4±2.6)%和(2.5±0.2)%;单纯东南亚型-HPFH 杂合子地贫患者分别为(75.0±5.3)fL 、(25.5±1.8)pg 、(23.8±5.2)%和(3.9±0.8)%;单纯台湾型杂合子地贫患者分别为(64.7±4.1)fL 、(20.2±2.1)pg 、(8.0±5.2)%和(6.4±1.1)%,3种地贫患者各项指标差异均有统计学意义(P <0.05)。


遗传疾病的遗传修饰与表观遗传调控遗传疾病是由基因异常引起的疾病,它们可以通过遗传修饰和表观遗传调控等机制来影响疾病的发生和发展。
本文将探讨遗传疾病中的遗传修饰和表观遗传调控两个重要角色。
一、遗传修饰遗传修饰是指在基因组水平上对基因表达进行调控的一种机制。
它可以通过改变基因序列中的DNA碱基,如突变、插入或缺失等,来影响基因产物的结构和功能。
这些基因突变可以直接导致疾病的发生,或者使个体对疾病易感。
例如,囊性纤维化病(Cystic Fibrosis,CF)是一种常见的遗传疾病,由CFTR基因突变引起。
这个基因编码一种离子通道蛋白,负责细胞膜的离子交换。
突变会导致CFTR蛋白质结构异常,进而影响离子通道的功能,导致黏液在肺部和胰腺等器官中大量积聚,引发疾病症状。
除了DNA序列突变外,基因的DNA甲基化和组蛋白修饰等也是遗传修饰的重要方式。
DNA甲基化是一种通过在DNA分子中添加甲基基团来改变基因表达的机制。
这种修饰方式可以导致某些基因的静默,从而影响特定功能或特征的表达。
二、表观遗传调控表观遗传调控是指在基因表达过程中对基因活性和表达进行调节的一种机制。
这种调控不涉及DNA序列的改变,而是通过化学修饰和染色质结构的组织状态改变等机制来实现。
表观遗传调控主要包括DNA甲基化、组蛋白修饰和非编码RNA等方式。
DNA甲基化已在前文中介绍过,它是通过加在DNA分子上的甲基基团来改变基因表达。
组蛋白修饰则是通过改变染色质上的蛋白质修饰方式,如酶促修饰、翻译后修饰和蛋白质相互作用等,来调节染色质结构和基因表达。
此外,非编码RNA也在表观遗传调控中起到重要作用。
非编码RNA是一类不编码蛋白质的RNA分子,在细胞过程中发挥多种调控功能。
它们可以通过与mRNA相互作用,调节基因表达、转录和翻译过程。
研究发现,一些非编码RNA在遗传疾病的发生中起到关键作用,如某些癌症和神经退行性疾病。
三、遗传修饰与表观遗传调控的关系遗传修饰和表观遗传调控在遗传疾病的发生和发展过程中常常相互作用。


基因突变导致的遗传性疾病表型与机制分析遗传性疾病作为一类由基因突变导致的常见疾病,近年来引起了广泛的关注。
在这篇文章里,我们将重点讨论基因突变所导致的遗传性疾病表型和机制,并探讨一些未来的治疗方法。
一、遗传性疾病表型遗传性疾病表型指的是由基因突变所引起的外显或隐性表现。
基因突变可以发生在体细胞或生殖细胞中,前者只会影响个体自身,而后者则可以被遗传到下一代。
举例来说,地中海贫血是一种遗传性疾病,主要由β-珠蛋白基因上的突变引起。
患者因此产生缺少正常血红蛋白的异常血红蛋白,导致贫血、溶血性危机和肝胆功能障碍等临床表现。
这是一种自显性疾病,也就是说只需有一个突变的基因就足以导致表型的发生。
另一方面,囊性纤维化是一种常见的常染色体隐性遗传疾病,由CFTR基因突变引起。
CFTR基因编码膜通道蛋白,患者突变后CFTR蛋白结构和功能异常,阻碍氯离子的转运,导致黏液过量和炎症等临床表现。
二、遗传性疾病机制基因突变会引起蛋白质结构和功能的改变,从而影响细胞、组织和器官的正常生理活动。
在患者中,这种影响会导致不同的病理变化和不同的临床表现。
在地中海贫血中,β-珠蛋白基因突变引起了珠蛋白基因链的异常拼接,导致异常珠蛋白的形成。
这种异常珠蛋白会聚集在红细胞内部,影响了红细胞形态和机能,导致贫血等表现。
在囊性纤维化中,CFTR基因突变导致了细胞内的钠离子、水分和黏液的输送异常,从而导致黏液分泌增加和器官功能障碍。
三、治疗方法目前,针对遗传性疾病的治疗方法主要包括基因治疗和细胞治疗。
基因治疗的目的在于通过基因替换、基因编辑和基因静默等手段,矫正患者的突变基因,在根本上解决疾病的发生。
而细胞治疗则主要是通过细胞移植等手段,将健康的细胞引入患者体内,从而提高患者的免疫力和生理功能。
比如,对于苯丙酮尿症这种遗传性代谢疾病,科学家们已经探索出了利用病毒载体导入正常PAH基因的方法,并成功治疗了一些患者。
而对于类似囊性纤维化这类基因缺陷较多、表型较多样的疾病,则需要更为复杂和精细的治疗方法。
遗传学中的基因修饰与表观遗传在遗传学中,基因是最基本的遗传因素,也是影响生物性状的核心因素。
但是,基因虽重要,但由于生物体内部复杂多样的生理环境和外部环境的影响,导致同一基因在不同物种中或不同个体中表现不同,表现为不同的性状和功能。
这就是因为基因并不是僵化的存在,而是随环境的变化而在表达上发生了改变。
基因修饰和表观遗传是影响基因表达的两大因素。
基因修饰是指基因发生突变或者在基因水平产生一些变化,从而影响基因表达。
基因修饰和遗传多一定程度上相同,但更多的是在表现上有所不同。
基因编码的酶会引起某些化学分子与DNA发生反应。
在这个过程中,化学分子加在DNA上,使它更易于或更难于读取。
基因修饰可以是加上分子或把某部分DNA切掉来增强或减弱基因表达。
与基因修饰不同的是,表观遗传指的是细胞核内染色体上的一些非编码序列的修饰或染色质的紧密度及其组成的调整,从而影响基因表达。
这些调整可以影响基因读码区外的表达,使得代际遗传差异得以延续。
基因修饰和表观遗传共同作用,共同构成了遗传学研究领域中基因表达和遗传变异的核心效应。
我们可以通过基因修饰和表观遗传的方式来调整某些特定基因的表达,从而达到预期的目的。
对生物体的研究更加深入,可以为我们提供对疾病的治疗和认识更多的可能,这是很有意义和重要的。
总之,基因修饰和表观遗传是在遗传学研究领域中常见的现象。
这两个过程在不同组织和细胞类型之间有所不同,其解析级别从基因序列、染色质、细胞核组和整个个体范围都可能存在多种研究领域。
随着生物技术的不断发展和技术的应用,有望为人类疾病治疗和认识提供更有效和更广泛的研究手段。
α地中海贫血基因型
α地中海贫血是异质性遗传性溶血性贫血,在各个类型的地中海贫血中,其发病率仅次于β地中海贫血。
以下是α地中海贫血基因型:
1. 静止型:正常人从父母双方各继承2个α珠蛋白基因(αα/αα),若继承的基因有一个或一个以上发生缺陷,可导致α珠蛋白链合成受到部分或完全抑制,引起α地中海贫血。
aa/aa表示拥有4个a珠蛋白基因,是正常的基因型。
2. 轻型:杂合子的静止型和轻型患者无症状或仅有轻微的血液学表现。
3. 中间型:表现为慢性溶血和脾大(临床特点)、轻度无效造血和血红蛋白合成总体减少而产生的小细胞低色素性红细胞和靶形红细胞(血象特点)以及血液HbF不增高、HbA2与HbA正常或减低和HbH与Hb Bart(复合杂合子的中间型和纯合子的重型)的遗传性溶血性贫血。
4. 重型:纯合子的重型患者可能出现严重的症状,包括出生后即出现严重贫血、黄疸和肝脾肿大等症状。
以上内容仅供参考,具体情况还需要根据患者的具体情况进行判断。
如有疑问,建议及时就医,寻求专业医生的帮助。