遗传性毛细血管扩张症(陆道培)
- 格式:doc
- 大小:17.00 KB
- 文档页数:3

遗传性出血性毛细血管扩张症的临床诊疗摘要】目的讨论遗传性出血性毛细血管扩张症诊治。
方法对遗传性出血性毛细血管扩张症进行诊治。
结论需反复输血者,注意预防乙型肝炎。
有双盲试验证明预防性用雌性激素可以减少严重出血者输血需要。
大多病人需长期使用铁剂,以补充粘膜反复出血所丧失的铁,某些患者需消化道外补铁。
【关键词】遗传性出血性毛细血管扩张症诊断与治疗遗传性出血性毛细血管扩张症(heredi-tary hemorrhagic telangiectasis HHT,又称 Rendu-Osier-Weber病)是一种遗传性皮肤及黏膜血管畸形的疾病。
临床上以反复出血及皮肤、黏膜毛细血管扩张为主要特征。
部分病人在内脏形成血管瘤或动静脉瘘,本病有15%合并肺动静脉瘘,肺动静脉瘘患者半数有HHT。
下面将遗传性出血性毛细血管扩张症的临床诊断与治疗报告如下。
1 病因、发病机理及病理本病为常染色体显性遗传,外显率约80%。
男女之比为1.5:1。
本病的发病机制尚未阐明。
其基本病变为全身毛细血管的局部扩张,并可形成血管瘤。
可能与先天性小动脉肌层薄弱或血管壁缺乏弹力纤维有关。
肺动静瘘造成右向左的分流。
少量分流在运动月才出现低氧血症。
较大的分流,即使吸纯自也不能完全纠正低氧血症。
分流量达目20%-30%或30%以上则症状明显。
2 临床表现2.1症状和体征多数成年患者在40-46岁才出现症状,幼年期发病的患者月在幼年期突然发生肺功能衰竭。
肝、脑、肾等器官也可发生血管瘤或动静脉瘘。
最常见的症状是呼吸困难及气短。
初为运动后呼吸困难,以后静息时也可出现呼吸困难。
直立或坐位使气短加重,平卧则减轻,称为直立型气短。
可能系直立时,地心引力使通过动静脉瘘的肺血流量增加,加重动脉的低氧血症。
吸氧可减轻缺氧症状,但患者仍有换气过度。
咯血亦较常见,也有大量咯血,但很少危及生命。
也可出现紫绀、杵状指、鼻衄等。
胸部病变处可闻及收缩期血管性杂音,吸气时尤甚。
病变破裂至胸腔可引起血胸。

奥氏征病理解释奥氏征(Osler-Weber-Rendu disease)又称为遗传性出血性毛细血管扩张症(Hereditary Hemorrhagic Telangiectasia,HHT),是一种遗传性疾病,主要特征是皮肤和黏膜上广泛分布的毛细血管扩张和脆性,导致出血。
本文将对奥氏征进行详细解释,包括其疾病特征、遗传模式、发病机制和临床表现等方面。
1. 疾病特征奥氏征是由于毛细血管稀疏、扩张和畸形导致的血管出血性疾病。
它遗传方式为常染色体显性遗传,但也有可能由于新生变异引起。
疾病患者常见的特征是皮肤和黏膜上出现散在或成群的红色或紫红色的扩张血管瘤,称为毛细血管扩张。
这些扩张血管瘤往往出血不止,导致患者反复出血和贫血。
2. 遗传模式奥氏征遗传方式为常染色体显性,而且在临床上呈不完全穿透性。
这意味着一个患有奥氏征的父母会将该病的遗传风险平均传递给他们的孩子,但不是所有的子女都会表现出该疾病。
即使是同一个家庭中,患病程度也可能有所不同。
3. 发病机制奥氏征的发病机制与内源性血管形成和修复机制的异常有关。
近年来的研究发现,该病的发病基因主要与血管内皮生长因子(Endoglin)和活性氧化物代谢酶(SMAD4)等相关。
异常的遗传基因导致血管内皮细胞的异常增殖和分化,进而引起血管扩张、脆性和出血的现象。
4. 临床表现奥氏征的临床表现多样化,常见症状包括鼻出血、呼吸道出血、消化道出血等。
此外,患者还可能出现皮肤瘀斑、手指末梢皮肤细胞增生、关节疼痛和慢性贫血等症状。
临床表现的严重程度和部位可以因个体差异而不同。
5. 诊断和治疗奥氏征的诊断主要依靠病史、临床表现和相关的遗传学研究。
血管造影、超声心动图和基因检测等技术也有助于确诊。
对于轻度病例,通常只需进行观察和管理。
对于严重病例,如有大量出血或临床症状对患者生活质量造成严重影响,可能需要进行手术治疗或药物干预。
结论:奥氏征是一种遗传性血管疾病,其主要特征是皮肤和黏膜上的毛细血管扩张和脆性,导致出血。

遗传性出血性毛细血管扩张症的研究进展遗传性出血性毛细血管扩张症是一种常染色体显性遗传疾病,以出血和血管扩张为主要表现。
其分子学发病机制为ENG和ALK1基因突变,临床症状主要表现为鼻出血、毛细血管扩张和内脏血管扩张。
遗传性出血性毛细血管扩张症的诊断依据临床表现和阳性家族史,临床上治疗以对症治疗和预防为主。
标签:遗传性出血性毛细血管扩张症;发病机制;临床表现;诊治遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)也称Osler-Rendu-Weber病,是一种常染色体显性遗传性血管发育异常型疾病,以出血和血管扩张为主要表现。
HHT以往一直被认为是一种少见的对患者无明显不利影响的疾病,然而,目前人们认识到HHT的发生率要比原来想象的高很多,且由于脑和肺脏病变会导致严重的致残率和致死率[1],因此,充分认识和学习HHT对治疗该疾病和预防严重并发症有很大益处。
1 病理生理学机制本病的病理学基础是毛细血管扩张和动静脉畸形。
毛细血管扩张多发生于口、鼻、胃肠道、皮肤及手指等部位,动静脉畸形多发生于胃肠道、肺、脑及肝脏等部位。
轻微病变表现为毛细血管后静脉局部出现扩张。
严重病变者血管出现显著扩张和扭曲,管壁由多层平滑肌组成而没有弹力纤维,且扩张的静脉常常与扩张的动脉直接相连。
2 分子遗传学发病机制目前,HHT的发生主要与两条染色体上的基因位点突变有关。
位于9号染色体的Endoglin(ENG)基因突变引起了1型HHT(HHT1)[2],位于12号染色体的活化素受体样激酶1(activin receptor-like kinase 1,ALK1)基因突变引起了2型HHT(HHT2)[3]。
ENG基因定位于9q33-34.1[2],ALK1基因定位于12q11-14[3]。
ENG和ALK1均编码转化生长因子β(transforming growth factory-β,TGF-β)家族的受体蛋白:ENG蛋白和ALK1蛋白,这两种受体蛋白主要在血管内皮细胞表面表达。



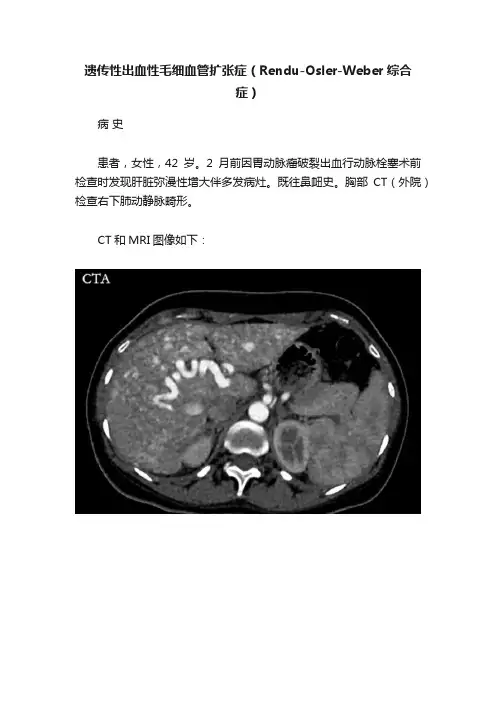
遗传性出血性毛细血管扩张症(Rendu-Osler-Weber综合症)病史患者,女性,42岁。
2月前因胃动脉瘤破裂出血行动脉栓塞术前检查时发现肝脏弥漫性增大伴多发病灶。
既往鼻衄史。
胸部CT(外院)检查右下肺动静脉畸形。
CT和MRI图像如下:影像表现CTA:肝总动脉、肝固有动脉、肝内动脉分支增粗,走行迂曲,肝总动脉直径约12mm,肝内见多发迂曲小血管影。
三支肝静脉动脉期早显,副肝右静脉显影(MIP)。
MRI:肝内多发大小不等T1稍低信号、T2稍高信号结节影,增强后持续明显强化,肝脏实质强化不均匀。
肝内血管影增粗迂曲,门脉系统及肝脏静脉早显。
基因诊断符合遗传性出血性毛细血管扩张症在患者的测序数据中,发现ACVRL1基因的上述已知致病突变。
ACVRL1基因是遗传性出血性毛细血管扩张症(Telangiectasis, hereditary hemorrhagic, type2, HHT2)[MIM:600376]的致病基因。
检测结论:检测到可以解释患者临床表型的致病突变讨论遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)又称Rendu-Osler-Weber综合症,是一种常染色体显性遗传性疾病,发病率约1/10000~1/5000。
HHT主要病理改变为皮肤、粘膜或内脏毛细血管、小动脉及小静脉管壁的结构异常,病变处血管壁菲薄且缺乏弹力纤维和平滑肌,导致病变处毛细血管扩张、动静脉畸形、动脉瘤形成,严重时可引起出血。
HHT血管变异本身无脏器器质性病变,早期大部分患者无相应临床症状。
鼻衄是HHT 最常见的症状,50%的患者在10岁之前发病,约95%的患者最终进展为反复发作性鼻出血。
皮肤黏膜的毛细血管扩张是最常见的体征,尤其于手部、脸部、嘴唇等特定部位见点样至大头针针帽大小非弥漫性的多发病灶,通常在20岁以后开始出现,病程后期几乎100% 的患者有此体征。

遗传性毛细血管扩张症(陆道培)毛细血管遗传性毛细血管扩张症(hereditaryhemorrhagictelangiectasis)是遗传性血管壁结构异常所致的疾病,患者部分毛细血管、小血管壁变薄,仅由一层内皮细胞组成,周围缺乏结缔组织支持,以致局部血管扩张,扭曲。
常见于口腔、鼻粘膜、手掌、指甲床和耳部及消化道。
病变呈针尖样、斑点状或斑片状、小结节状,也可呈血管瘤样或蜘蛛痣样,可高出皮肤表面,加压后消失,用玻片轻压有时可见小动脉搏动。
疾病简介遗传性毛细血管扩张症(hereditaryhemorrhagictelangiectass)是遗传性血管壁结构异常所致的疾病,患者部分毛细血管、小血管壁变薄,仅由一层内皮细胞组成,周围缺乏结缔组织支持,以致局部血管扩张,扭曲。
常见于口腔、鼻粘膜、手掌、指甲床和耳部及消化道。
病变呈针尖样、斑点状或斑片状、小结节状,也可呈血管瘤样或蜘蛛痣样,可高出皮肤表面,加压后消失,用玻片轻压有时可见小动脉搏动。
临床上以病变部位自发性或轻伤时反复出血为特征,多表现为鼻衄、牙龈出血。
内脏出血以呕血、黑便为多见,也可有咯血、血尿、月经过多、眼底或颅内出血等。
出血症状可在幼年出现。
实验室检查大多正常,出血严重者可有贫血。
甲床毛细血管镜检可有血管襻异常扩张。
诊断与蜘蛛痣和红痣相鉴别。
发病机制遗传性毛细血管扩张症(HHT)和家族性肺动脉高压均为编码转化生长因子(TGF) β受体的蛋白[包括活化素受体激酶1 (ALK 1)、内皮因子和骨形态发生蛋白2(BMPR 2)]基因突变引起的血管系统病变。
内皮细胞表面TGFⅡ型受体(如BMPR 2)在Ⅲ型受体(如内皮因子)辅助作用下,与Ⅰ型受体(如ALK 1)结合形成跨膜复合物,激活Ⅰ型受体激酶域,促进其胞内蛋白磷酸化,激活下游Smad信号,进入胞核促进基因转录,调节血管的分化和增殖。
该通路中的任何组分,包括ALK 1、BMPR 2和内皮因子突变,均可能与肺动脉高压有关。


先天性毛细血管扩张性大理石样皮肤2例并文献复习王反玲许红波王源(厦门长庚医院,福建厦门361028)定,150例中有148例达到有效浓度,只有2例未达到有效浓度,而传统痰盂因未加盖受到外界很多因素的影响,尤其是环境因素,150例中有12例未达到有效浓度,差异有统计学意义(P <0.05)。
改良后痰盂在传统痰盂基础上加盖加提手,加盖可以防止消毒剂不被挥发,更能保证消毒剂的有效浓度,还能有效杀灭结核杆菌,切断传播途径,防止交叉传播;加提手方便患者拿放,使用时打开桶盖,吐好痰液然后及时盖好桶盖,提手的高度稍微高于床沿,无论患者是卧位还是坐位,都能及时打开桶盖,方便吐痰。
而且使用改良痰盂后,病房内消毒液刺激异味明显好转,空气得到明显改善。
使用过程中消毒液的浓度保持在稳定状态,且避免了空气中的粉尘、微生物等污染消毒液。
从经济学方面看,改良后痰盂加盖加提手并不增加患者经济费用,其他血液、体液传染患者的胸水、腹水等也可用改良后痰盂作为容器消毒处理。
总之,改良后痰盂加盖加提手,既能保证消毒剂的有效浓度,又能杀菌,防止交叉感染,且费用不变,使用方便。
参考文献[1]曲显恩.含氯消毒剂的性能与应用[J].中国氯碱,2005,9(1):19-23.[2]GRIFFITHS P A ,BABB J R ,FRAISE A P.Mycobacterium terrae :A potential surrogate for mycobacterim tuberculosis in a standard disinfectant test[J].J Hos Infect ,1998(38):183.[3]王晓辉,张朝武,陈昭斌,等.4株分枝杆菌对含氯消毒剂抗力的比较[J].中国消毒学杂志,2002,19(1):12-15.(收稿日期:2019-11-17)DOI :10.19435/j.1672-1721.2020.04.076先天性毛细血管扩张性大理石样皮肤(cutis marmoratatelangiectatica congenita ,CMTC )是一种罕见的先天性血管畸形,又称为van Lohuizen 综合征、先天性泛发性静脉扩张症,由vanLohuizen 于1922年首次报道[1],主要表现为出生时即发生广泛或局限性的青灰色网状斑点。

遗传性毛细血管扩张症病因病理、临床表现及诊断要点遗传性毛细血管扩张症是血管发育异常所引起的出血性疾病。
本病是一种波及全身血管的家族性疾病,主要特征为小动脉、小静脉及毛细血管的局限性扩张和纡曲,导致异常出血倾向,临床常见为同一部位反复出血或轻微受伤后出血不止,以鼻衄最常见。
本病为染色体显性遗传,男女均可发病,该病为终生疾病,纯合子者的死亡率甚高。
1896年Rendu首次以反复鼻衄报道本病。
继之,他人又发现本病为家族性遗传性疾病。
国内1961年发表个案报道,1979年发现三个家系的大宗病例。
国内报告本病的发病率为1~2/10万。
本病属中医学“血证”范畴。
【病因病理】一、西医1.病因 本病以常染色体显性方式遗传,外显率比较高。
男女皆可发病,但女性的出血倾向常比男性轻。
双亲均可遗传。
缺乏阳性家族史并不否定遗传性出血性毛细血管扩张症的诊断,有人认为这类病是由于基因突变或病史资料不完全。
在诊断本病时应注意隔代遗传现象。
2.病理 本病病理变化的特征性病变是紧靠粘膜下或真皮内的扩张纡曲而菲薄易碎的血管团,往往仅由单层内皮细胞所组成,病变血管周围只有一些疏松的结缔组织,一般缺乏平滑肌结构或弹性纤维。
因而患有病变的毛细血管、小静脉和小动脉缺乏收缩能力,并容易脆破。
病变的血管多呈血管瘤样改变,病变严重的血管往往呈动静脉瘘样。
二、中医本病的产生,多由先天禀赋不足,肝肾阴亏,脾虚失统所致。
综合病因、病机主要有以下几个方面。
1.禀赋不足 肾为先天之本,元气之根,是促进人体生长发育的原动力。
肾气旺盛,才会身体健壮。
若禀赋不足,真元亏损,真阴化生不足而致阴虚火旺,迫血妄行,发为本病。
2.肝肾阴亏 平素肝旺或肝肾阴亏,肝火灼阴,阴亏火旺,火旺络伤,血溢脉外,乃生此病。
3.脾虚失统 劳倦过度,或饮食伤脾,损伤脾气,统摄无权,血无所归而妄行,溢出脉道而成本病。
由此可知,先天禀赋不足及后天失养是导致本病的主要原因。
肾亏火旺、脾虚失统为其主要病理机转。
遗传性出血性毛细血管扩张症是怎么回事
*导读:本文向您详细介绍遗传性出血性毛细血管扩张症的病理病因,遗传性出血性毛细血管扩张症主要是由什么原因引起的。
*一、遗传性出血性毛细血管扩张症病因
*一、病因:
1.遗传因素(90%):
本病为常染色体显性遗传性疾病,男女均可患病,均可遗传,病变部位在血管壁,表现为毛细血管扩张,动静脉畸形和动脉瘤,血管壁变薄,弹力纤维缺乏,平滑肌缺乏,毛细血管壁和小动脉壁仅由一层内皮细胞组成,血管迂曲或扩张,有时仅有的内皮细胞发生退行性变,内皮细胞连接缺损,病变血管可因轻微的外力,或血管内血流压力作用即可发生破裂而出血。
*温馨提示:以上就是对于遗传性出血性毛细血管扩张症病因,遗传性出血性毛细血管扩张症是由什么原因引起的相关内容叙述,更多有关遗传性出血性毛细血管扩张症方面的知识,请继续关注疾病库,或者在站内搜索“遗传性出血性毛细血管扩张症”找到更多扩展内容,希望以上内容可以帮助到您!。
如对您有帮助,可购买打赏,谢谢
生活常识分享遗传性毛细血管扩张临床表现
导语:遗传性毛细血管扩张是一种人体血管疾病,会表现在人体皮肤表面上,严重的患者会造成一些血液疾病,遗传性毛细血管扩张患者十分痛苦,皮肤上
遗传性毛细血管扩张是一种人体血管疾病,会表现在人体皮肤表面上,严重的患者会造成一些血液疾病,遗传性毛细血管扩张患者十分痛苦,皮肤上也会出现一些红色的血丝,用肉眼就可以看到血管,皮肤上也会出现一些疙瘩,下面让我们一起来了解一下遗传性毛细血管扩张。
临床表现:
多在20~30岁之间发病,部分在儿童期即可发病。
最突出的症状是受累血管破裂出血,常在同一部位反复出血。
儿童期多见鼻衄,到青少年期鼻衄渐趋好转,而内脏出血机会增加,以胃肠道出血最多见,其他可有咯血、血尿、眼底出血、月经过多、蛛网膜下腔出血等。
肝脏受累,因流经肝动静脉瘘的血流量增多而出现肝肿大,可有肝区疼痛及一定程度的压痛,局部有时可触及一搏动性肿块,触之有震颤,能闻及连续性血管杂音。
动-静脉瘘的分流可产生高动力循环状态,并可产生高排量充血性心力衰竭,可因肺的动静脉瘘而引起低氧血症、继发性红细胞增多症。
慢性失血或频繁而大量出血可致缺铁性贫血。
毛细血管先天性畸形即先天性毛细血管壁薄弱,以致不能收缩。
本病多自出生时或生后不久发生于面部、颈部和枕后、头皮部。
可单侧,散发,亦可双侧,多发。
最初皮肤或粘膜上有一个大小不一,淡红色或暗红,或紫红色皮损,自针尖大小至一个肢体或半侧躯干,哭闹后颜色加深,界限清楚,形状各异,不高出皮肤,局部则较高,压迫后,部分或全部退色,表面光滑。
随年龄增长,如儿童或青壮年有可能在。
遗传性出血性毛细血管扩张症的治疗*导读:遗传性出血性毛细血管扩张症是常染色体显性遗传性血管发育异常的一种疾病。
1864年该病由SUTTON首例报道。
1896年RENDU对该病进行了较详细地论述。
……遗传性出血性毛细血管扩张症是常染色体显性遗传性血管发育异常的一种疾病。
1864 年该病由SUTTON 首例报道。
1896 年RENDU 对该病进行了较详细地论述。
1901 年OSLER 报道该病的家族性及其临床特征。
以后,WEBER 也阐述了该病例。
因此,该病用三者的名字命名,也称为Osler- Rendu-Weber 病。
1909 年Hanes 以彩色图示颇为全面地讨论了该病,并把该病命名为遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia,HHT)。
HHT 只能对症和支持治疗,目前尚无特效治疗措施。
止血应尽可能用非创伤性手段。
手术止血或其他原因接受止血,应特别注意扩张的毛细血管发生术中和术后出血。
1 鼻出血对鼻出血特别是如果患者感到频繁或持久干扰正常的活动或危及健康时,应考虑适当的介入疗法。
患者每天应用鼻腔润滑剂进行湿润是有用处的。
建议患者避免用力擤鼻子、抬重物、要防止大便燥结以避免大便时太用力、不要用指头抠鼻垢。
对HHT 的鼻出血,临床对一些治疗进行了评估。
为控制轻度鼻出血减轻鼻出血,应用激光切除疗法是最有效的介入疗法。
具有严重的鼻出血,应有熟练鼻中隔皮肤成形术的耳鼻喉科专家进行厚皮移植,其具有好的疗效。
具有治疗HHT 患者经验的大部分耳鼻喉科专家建议避免使用电的和化学烧灼术治疗,对于许多复发性鼻出血的治疗使用经导管栓塞疗法。
2 胃肠道出血如果经过充分的铁剂治疗,血红蛋白浓度仍不能维持在正常范围内,就应该考虑其他方法。
为了确定出血部位及其类型( 如:毛细血管扩张、AVM)、可以使用内窥镜、肠系膜及腹腔血管造影术、放射性核素。
加热器探针内窥镜的应用、双囊、或激光是主要依靠的局部治疗方法。
遗传性毛细血管扩张症(陆道培)
毛细血管遗传性毛细血管扩张症(hereditaryhemorrhagictelangiectasis)是遗传性血管壁结构异常所致的疾病,患者部分毛细血管、小血管壁变薄,仅由一层内皮细胞组成,周围缺乏结缔组织支持,以致局部血管扩张,扭曲。
常见于口腔、鼻粘膜、手掌、指甲床和耳部及消化道。
病变呈针尖样、斑点状或斑片状、小结节状,也可呈血管瘤样或蜘蛛痣样,可高出皮肤表面,加压后消失,用玻片轻压有时可见小动脉搏动。
疾病简介
遗传性毛细血管扩张症(hereditaryhemorrhagictelangiectass)是遗传性血管壁结构异常所致的疾病,患者部分毛细血管、小血管壁变薄,仅由一层内皮细胞组成,周围缺乏结缔组织支持,以致局部血管扩张,扭曲。
常见于口腔、鼻粘膜、手掌、指甲床和耳部及消化道。
病变呈针尖样、斑点状或斑片状、小结节状,也可呈血管瘤样或蜘蛛痣样,可高出皮肤表面,加压后消失,用玻片轻压有时可见小动脉搏动。
临床上以病变部位自发性或轻伤时反复出血为特征,多表现为鼻衄、牙龈出血。
内脏出血以呕血、黑便为多见,也可有咯血、血尿、月经过多、眼底或颅内出血等。
出血症状可在幼年出现。
实验室检查大多正常,出血严重者可有贫血。
甲床毛细血管镜检可有血管襻异常扩张。
诊断与蜘蛛痣和红痣相鉴别。
发病机制
遗传性毛细血管扩张症(HHT)和家族性肺动脉高压均为编码转化生长因子(TGF) β受体的蛋白[包括活化素受体激酶1 (ALK 1)、内皮因子和骨形态发生蛋白2(BMPR 2)]基因突变引起的血管系统病变。
内皮细胞表面TGFⅡ型受体(如BMPR 2)在Ⅲ型受体(如内皮因子)辅助作用下,与Ⅰ型受体(如ALK 1)结合形成跨膜复合物,激活Ⅰ型受体激酶域,促进其胞内蛋白磷酸化,激活下游Smad信号,进入胞核促进基因转录,调节血管的分化和增殖。
该通路中的任何组分,包括ALK 1、BMPR 2和内皮因子突变,均可能与肺动脉高压有关。
根据分子遗传学机制的不同,HHT分为3型。
HHT 1型通常为内皮因子基因突变所致,HHT 2型和HHT 3型分别为ALK 1和Smad 4基因突变所致。
既往研究显示,HHT相关肺动脉高压主要发生于1型HHT,表现为肺动脉阻力增高,肺动脉压力增加,心输出量下降。
临床表现
多在20~30岁之间发病,部分在儿童期即可发病。
最突出的症状是受累血管破裂出血,常在同一部位反复出血。
儿童期多见鼻衄,到青少年期鼻衄渐趋好转,而内脏出毛细血管扩张血机会增加,以胃肠道出血最多见,其他可有咯血、血尿、眼底出血、月经过多、蛛网膜下腔出血等. 肝脏受累,因流经肝动静脉瘘的血流量增多而出现肝肿大,可有肝区疼痛及一定程度的压痛,局部有时可触及一搏动性肿块,触之有震颤,能闻及连续性血管杂音。
动-静脉瘘的分流可产生高动力循环状态,并可产生高排量充血性心力衰竭,可因肺的动静脉瘘而引起低氧血症、继发性红细胞增多症。
慢性失血或频繁而大量出血可致缺铁性贫血。
毛细血管先天性畸形即先天性毛细血管壁薄弱,以致不能收缩。
本病多自出生时或生后不久发生于面部、颈部和枕后、头皮部。
可单侧,散发,亦可双侧,多发。
最初皮肤或粘膜上有一个大小不一,淡红色或暗红,或紫红色皮损,自针尖大小至一个肢体或半侧躯干,哭闹后颜色加深,界限清楚,形状各异,不高出皮肤,局部则较高,压迫后,部分或全部退色,表面光滑。
随年龄增长,如儿童或青壮年有可能在其上有症
状或结节状损害,多数发生在小腿和足部,可表现为疼痛性紫蓝色结节和斑块,尚可破溃。
本病为常染色体显性遗传性疾病,男女均可患病,父母均可遗传,常有家族史。
无特殊治疗方法,以对症治疗为主,浅表出血可用局部压迫止血,内脏出血处理较困难,必要时可手术缝合或切除病变或局部使用止血剂。
慢性失血性贫血可常规补充铁剂,出血多者需输血。
临床检查
毛细血管镜检查在病变部位可见小血管扩张扭曲,有时可见许多管壁菲薄的扩张血管聚集成较大的血管团。
内脏出血者在脏器局部可见相应的病变,如:胃肠道毛细血管扩张者内窥镜下可见胃肠道黏膜的点状血管扩张;肺内血管病变者胸片可见血管束与肺门相连的硬币样致密影。
组织病理学检查
本病的基本病理变化可见于全身各个部位,尤其是面部毛细血管、黏膜和内脏的毛细血管、小动脉及小静脉管壁结构异常,血管壁变得异常菲薄,有的部位仅有一层内皮细胞,外围包裹一层疏松结缔组织,缺乏正常血管壁的弹力纤维及平滑肌成分。
同时血管壁失去对交感神经和血管壁活性物质调节的反应能力,缺乏正常的舒缩功能,以至在血流的冲击下,病变部位的血管可发生结节状和瘤状扩张,严重时可形成动静脉瘘和动静脉瘤,可引起出血。
诊断原理
阳性家族史、毛细血管扩张及同部位的反复出血有助于诊断。
另外,由于患者血管壁脆弱,临床上束臂试验常阳性,并有出血时间延长。
同时,血管造影有确诊价值。
最近开始有DNA测试,有助诊断未发病的患者,预防严重的脑或肺出血。
若有肺动静脉瘘家族史,在青春期进行肺电脑扫描或脑核磁共振检查,将有助于诊断。
注意须与蜘蛛痣和红痣相鉴别。
阳性家族史、毛细血管扩张及同部位的反复出血。
由于血管壁的脆弱,临床上束臂试验常阳性,并有出血时间延长。
血管造影有确诊价值。
遗传性毛细血管扩张症容易与哪些疾病混淆?注意须与以下疾病相相鉴别:蜘蛛痣、红痣、小静脉扩张。
诊断标准
2000年国际HHT基金科学顾问委员会的诊断标准如下。
1. 鼻衄:反复、自发性鼻出血;
2. 毛细血管扩张:位于特征部位(如嘴唇、口腔、手指和鼻部)的多发毛细血管扩张;
3. 内脏损害:如胃肠毛细血管扩张(伴或不伴出血)、肺动静脉畸形、肝脏动静脉畸形、脑动静脉畸形和脊椎动静脉畸形;
4. 家族史:根据上述诊断,患者一级亲属中,至少有1位被诊断为HHT。
以上4项中,符合3项即可确诊HHT,符合2项则疑诊为HHT,如少于2项则诊断可能性不大。
治疗方法
大多患者无特殊治疗方法,主要为对症疗法。
但由于肺动静脉瘘危险,即使无症状,也应该彻底检查有没有此问题,以便做预防性栓塞治疗。
在未能栓塞消除肺动静脉瘘之前,任何手术或牙科治疗都应该加用预防性抗生素,以避免脑脓肿。
对可接触到的毛细血管扩张病变(如皮肤,或经内镜进入鼻或消化道),可直接压迫,也可行激光摘除术、手术缝合、切除病变或用局部止血剂等。
动静脉瘘可行外科切除或栓塞疗法。
需反复输血者,注意预防乙型肝炎。
有双盲试验证明
预防性用雌性激素可以减少严重出血者输血需要。
大多病人需长期使用铁剂,以补充粘膜反复出血所丧失的铁,某些患者需消化道外补铁(参见缺铁性贫血的治疗)。
肝动脉栓塞可用于治疗肝动静脉瘘。
β-受体阻滞剂可改善高动力循环状态,降低肝血流量,使分流量减少。
遗传性毛细血管扩张症治疗方法主要分为以下几个步骤:
1.止血。
体表出血以压迫止血为主,内脏出血者考虑用安络血以助小血管收缩,用垂体后叶素降低内脏血管内压力。
2.输血。
仅用于大量失血者,但不宜过量,避免血压过高而使出血难止。
3.补充铁剂。
适用于慢性失血性贫血患者。
4.其他。
肝动脉栓塞可用于治疗肝动静脉瘘。
β-受体阻滞剂可改善高动力循环状态,降低肝血流量,使分流量减少。