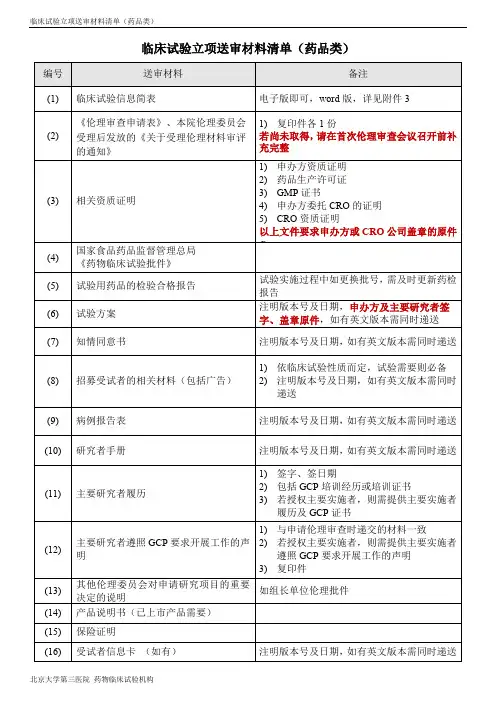
临床试验立项送审材料清单药品类
- 格式:doc
- 大小:40.00 KB
- 文档页数:2
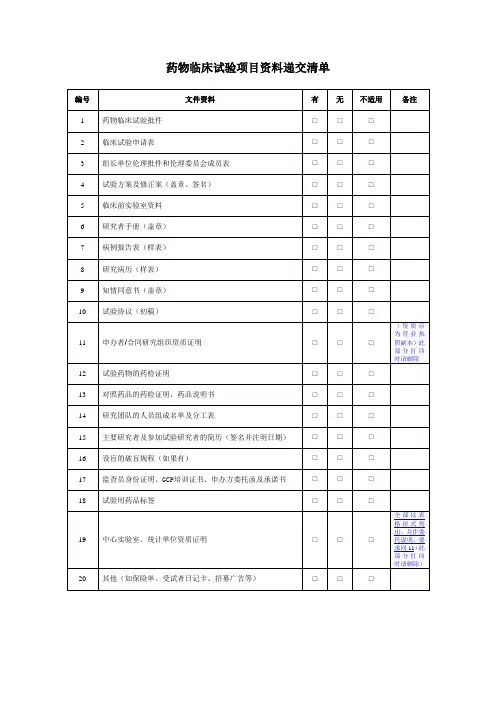

药物临床试验机构立项提交资料清单一、注意事项:1.机构立项资料递交信仅收取一份原件。
2.立项材料整体的第一页及每一部分的首页均要盖章,整体盖骑缝章。
如项目是CRO承担的,CRO的章也可以,但注明盖申办方原章的除外,不接受盖章后的彩色打印件。
3.《立项申请表》、《临床试验信息表》及《药物临床试验专业方案审核意见表及质量承诺书》在提交立项资料时一并递交,仅收取各一份原件由提交人盖章,此三份文件请勿列在立项资料目录中。
4.如递交立项资料目录中没有列出的文件,也需在文件上盖章,2页及2页以上的文件需盖齐缝章。
5.文件资料请用黑色、厚壳、双孔快劳夹装订,文件夹侧标使用统一的模板(应包含:院内项目编号、方案编号、项目名称、申办方名称、研究专业、主要研究者、批件号),模板参见附件1。
6.文件夹第一页均为《文件目录》,每项文件中间用带数字标识的隔页纸分隔。
7.请按立项申请表文件目录所列文件顺序排列各项文件,如提交《立项资料目录》中未列出的文件,请在立项申请表目录中补充完整。
文件夹中不包括的文件无需列入目录。
8.文件多的项目请准备多个“机构文件夹”,编号“①,②,③……”。
9.请在接待日,由负责项目监查的CRA将文件夹递交到机构办公室。
10.递交纸版文件资料的之前请发送一份填写完整的电子版立项文件至到机构办邮箱:(******)),立项及之后的内次备案资料均要求电子版资料内容与纸质版保持一致。
11.如立项时已有CRC的项目,请准备SMO公司资质证明、CRC中文版个人简历(并包括该CRC在我院所负责的所有项目名称、承担专业、项目院内编号、CRC 的联系电话和邮箱)至机构备案登记。
12.如在项目进行过程中更换监查员,请及时通知机构办,并递交人员变更通知函、新任监查员委托书/函、新任监查员GCP培训证书进行备案登记。
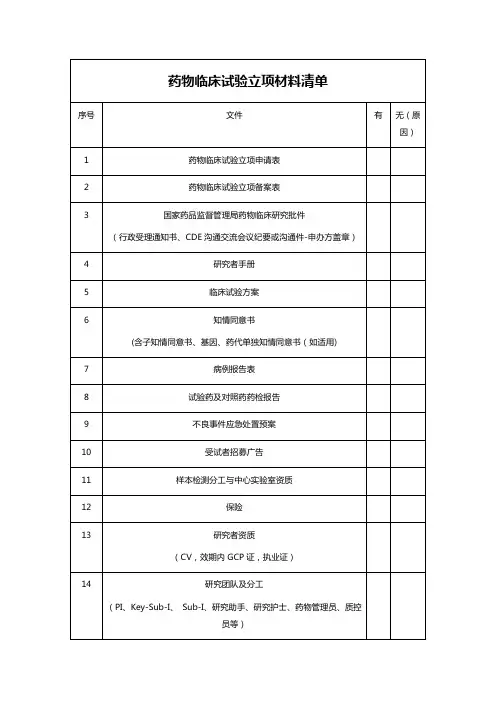

临床试验需提供资料列表清单药品类一、药品基本信息资料1、药品名称:包括通用名、商品名、化学名。
2、药品成分:列出药品的所有活性成分和非活性成分。
3、药品剂型与规格:说明药品的剂型(如片剂、胶囊、注射剂等)以及各种规格。
4、药品的生产工艺:包括原材料的来源、合成或提取过程、制剂工艺等。
5、药品的质量控制标准:包括各项质量指标的检测方法和限度。
二、药品研发相关资料1、前期研究报告:如药物的筛选、合成路线的确定、药理毒理的初步研究等。
2、药学研究报告:包括处方设计、稳定性研究、溶出度研究等。
3、药理毒理研究报告:涵盖药效学、药代动力学、急性毒性、长期毒性、致畸致癌致突变等试验的研究结果。
三、临床试验方案相关资料1、临床试验方案:详细描述试验的目的、设计、方法、纳入和排除标准、治疗方案、观察指标、随访时间等。
2、知情同意书:向受试者提供的关于试验的详细说明,包括试验的风险和受益,以获取受试者的自愿同意。
3、研究者手册:为研究者提供关于药品的全面信息,包括药品的药理作用、毒理作用、临床前研究结果、用法用量、不良反应等。
四、临床试验机构和研究者相关资料1、临床试验机构的资质证明:如医疗机构执业许可证、伦理委员会批件等。
2、研究者的资质证明:包括医师资格证书、执业证书、培训经历等。
3、研究者的简历和相关经验介绍。
五、药品生产和质量相关资料1、药品生产企业的资质证明:如药品生产许可证、GMP 证书等。
2、药品的生产批件或注册证。
3、药品的检验报告:包括出厂检验报告、抽检报告等。
4、药品的稳定性研究数据:包括长期稳定性和加速稳定性研究结果。
六、安全性相关资料1、药品的不良反应报告:包括已有的临床应用中的不良反应记录,以及动物试验中的不良反应观察结果。
2、风险控制计划:针对可能出现的风险制定的相应控制措施和应急预案。
七、统计学相关资料1、样本量计算依据:说明确定试验所需样本量的方法和依据。
2、统计分析计划:详细描述试验数据的统计分析方法和流程。

临床试验需提供资料列表清单药品类一、药品研发相关资料1、药品的化学、药学和生物学特性药物的化学结构、合成路线和纯度分析报告。
制剂的处方组成、制备工艺和质量控制标准。
药物的稳定性研究数据,包括影响因素试验、加速试验和长期试验结果。
2、非临床研究资料药理毒理研究报告,包括药效学、药代动力学和毒理学试验结果。
动物体内的安全性评价,如急性毒性、长期毒性、生殖毒性、遗传毒性等研究报告。
3、药品质量标准和检验报告拟定的药品质量标准草案,包括性状、鉴别、检查、含量测定等项目。
按照质量标准进行检验的检验报告,证明药品的质量符合拟定标准。
二、临床试验方案相关资料1、临床试验方案详细描述试验的目的、设计、方法、纳入和排除标准、治疗方案、观察指标、疗效评价标准、安全性评估等内容。
注明试验的分期(如 I 期、II 期、III 期等)和试验类型(如随机对照试验、开放试验等)。
2、研究者手册提供给研究者的关于药品的安全性、有效性、药理毒理等方面的详细信息,以帮助研究者更好地理解和开展试验。
3、知情同意书向受试者告知试验的相关信息,包括试验目的、方法、风险和受益等,确保受试者在充分知情的情况下自愿参加试验。
三、临床试验机构和研究者相关资料1、临床试验机构的资质证明医疗机构的执业许可证副本。
相关专业科室的设置和诊疗能力证明。
2、研究者的资质证明研究者的医师资格证书、医师执业证书。
研究者的履历和相关培训经历证明。
3、伦理委员会的审查文件伦理委员会的批件,证明试验方案和相关文件已经过伦理审查并获得批准。
四、临床试验受试者相关资料1、受试者的筛选和入选记录受试者的基本信息,如姓名、年龄、性别、诊断等。
筛选过程中的检查和检验结果,以确定受试者是否符合入选标准。
2、受试者的治疗记录每次给药的时间、剂量和途径。
受试者在试验期间的症状、体征、实验室检查结果等观察记录。
3、受试者的随访记录试验结束后的随访时间、内容和结果。
五、临床试验数据和统计分析相关资料1、临床试验数据管理计划描述数据的采集、录入、核查、存储和传输等流程和方法。


临床试验需提供资料列表清单药品类一、药品研发相关资料1、药品的化学、药学和生物学信息药物的化学结构、分子式、分子量等详细信息。
合成路线、制备工艺以及质量控制标准。
药物的稳定性研究报告,包括在不同条件下(如温度、湿度、光照等)的稳定性数据。
2、药效学和药代动力学研究资料药效学研究报告,说明药物在体内的作用机制、药效强度和持续时间等。
药代动力学研究报告,包括药物的吸收、分布、代谢和排泄情况。
药物相互作用的研究结果,尤其是与其他常用药物可能产生的相互作用。
3、毒理学研究资料急性毒性研究报告,包括动物的致死剂量、中毒症状等。
长期毒性研究报告,评估药物在长期使用情况下对器官功能的潜在影响。
特殊毒性研究报告,如致畸、致癌、致突变等试验结果。
二、临床试验方案相关资料1、临床试验方案详细的试验目的、设计类型(如平行组设计、交叉设计等)、研究人群、样本量计算依据。
试验药物的用法用量、给药途径和疗程安排。
主要疗效指标和次要疗效指标的定义和测量方法。
安全性评估的方法和指标,包括不良事件的记录和处理流程。
2、研究者手册药物的特性、作用机制、临床前研究结果等综合信息。
临床试验中的注意事项、可能的风险和应对措施。
药物的剂量调整原则和特殊情况的处理方法。
三、伦理审查相关资料1、伦理审查申请表填写试验的基本信息、研究者和申办者的相关情况。
对试验涉及的伦理问题进行初步说明。
2、知情同意书以通俗易懂的语言向受试者说明试验的目的、过程、风险和受益。
明确受试者的权利和义务,以及隐私保护措施。
提供受试者联系电话和咨询方式。
四、研究者和研究机构相关资料1、研究者简历主要研究者和参与研究者的教育背景、工作经历、专业资质和临床试验经验。
研究者在相关领域的研究成果和发表的论文。
2、研究机构资质证明研究机构的医疗机构执业许可证副本。
相关专业科室的设置和设备清单。
3、研究团队培训记录对研究人员进行临床试验方案、操作流程、安全保障等方面的培训记录。
五、试验用药品相关资料1、药品生产和质量控制文件药品生产企业的资质证明,如药品生产许可证。
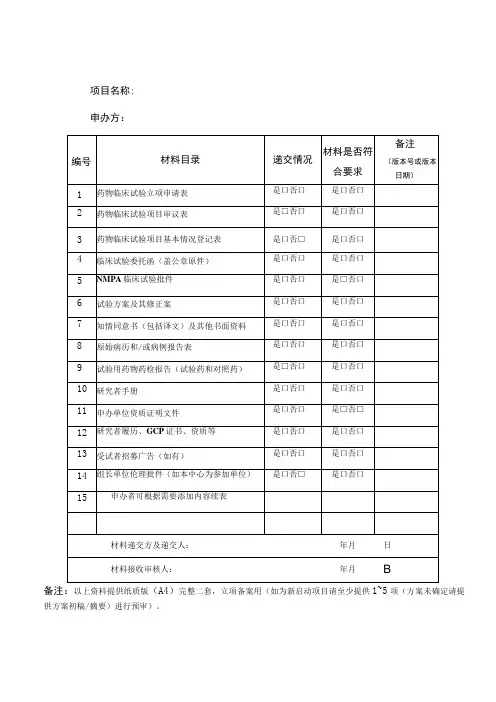
药物临床试验项目受理资料清单
(请以A4黑色快劳夹按如下顺序放置,用带序号的分隔页隔开,并做好目录及侧标)
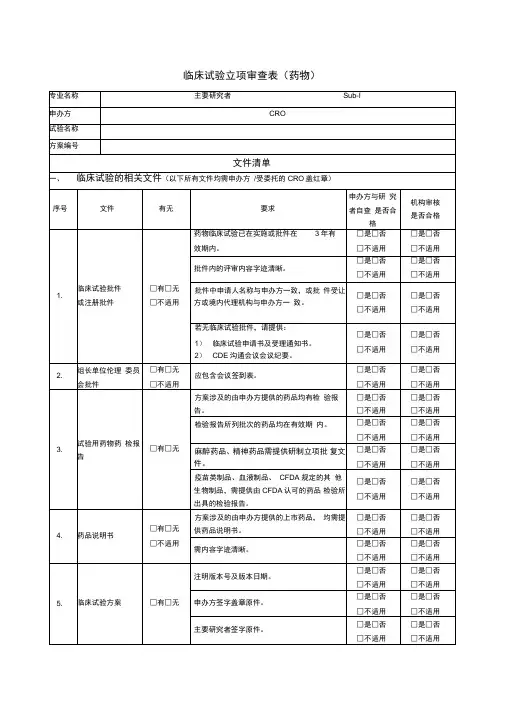
1.药物临床试验申请表(主要研究者签名签日期)
2.临床试验批件/受理通知书
3.组长单位伦理批件
4.临床试验委托书
5.临床试验方案(主要研究者签名签日期,标明版本号、版本日期)
6.知情同意书及相关材料(标明版本号、版本日期)
7.招募受试者相关材料(标明版本号、版本日期)
8.病例报告表(标明版本号、版本日期)
9.研究者手册(标明版本号、版本日期)
10.药品检验报告(试验药、对照药、安慰剂)
11.研究者履历及相关文件(履历需研究者签名签日期,GCP证书复印件)
12.申办方/CRO资质
13.申办方授权CRO的委托书(如有)
14.试验用药品标签
15.临床试验保险(如有)
16.实验室检测正常值范围
17.药物临床试验参加人员
18.其它需要审查的材料(如有)
盖章说明:
除了第1、11和17项文件外,其余文件均需加盖申办方公章。
盖章要求:首页、签字处、文件多于一页者加盖骑缝章。
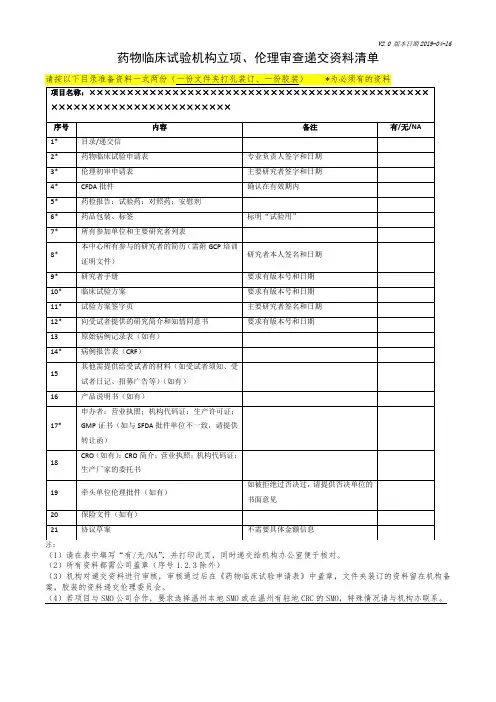
26 申办方/CRO营业执照□是□否
27 申办方对CRO的委托书或协议(如有)□是□否
28 申办方/CRO对CRA的委托书□是□否
□是□否29 CRA的派遣函、身份证复印件、学历证书、个人简历、GCP证
书等
30 无需办理“人类遗传资源审批申请书”承诺书(如适用)□是□否
31 方案讨论会的会议纪要□是□否31 其他与立项审查相关的材料□是□否
□是□否32 提供给受试者的文件,如调查问卷病、受试者日记卡等(如
适用)
备注:
1、以上所有材料需加盖红章,一式两份;装订要求:每一小
项放在一份文件保护袋中,再按照目录顺序要求放入三孔
文件夹中,用索引纸分开(参考如下:齐心TC530AB 文
件夹A4打孔夹背宽40mm 1.5寸3孔O型档案夹,蓝
色天猫
+%BF%D7+o%D0%CD%BC%D0+%C6%EB%D0%C4 。
3孔O
型夹齐心蓝色)。
2、将电子档或扫描件发邮箱:.。
临床试验立项递交资料清单(一)药物临床试验1. 递交文件目录(含所递交文件的清单,注明所用提交文件的版本号或日期)2. 临床试验评估表和立项申请表(下载区);3. 临床试验项目委托书;4. 国家食品药品监督管理总局(CFDA)药物临床试验批件;5. 组长单位伦理委员会批件(除本中心为组长单位免提供);6. 其他伦理委员会的批件或对申请研究项目的重要决定的说明,应提供以前否定结论的理由(如有请提供);7. 临床试验方案及其修正案(注明版本号及日期,外文资料的中文版,方案封面:各试验中心PI签名及日期);8. 知情同意书(注明版本号及日期,外文资料的中文版);9. 病例报告表(注明版本号及日期);10. 研究病历(注明版本号及日期);11. 研究者手册;12. 试验药物质检报告(必须加盖药品生产厂家红章);13.临床前实验室资料;14.试验用药品标签;15. 研究者履历表及相关文件(PI签名及日期);16. 申办方的资质证明(营业执照、生产许可证、GMP证书,税务登记证,组织机构代码证等复印件);17.CRO及SMO公司资质证明(如有);18. 监查员和CRC的法人委托书,身份证复印件,学历证明、GCP 培训证书及个人研究简历;19. 其他试验相关文件提供资料(如受试者须知、受试者日记卡、研究问卷表、招募广告、保险声明等)。
备注:1、以上所有材料需加盖红章,一式两份;装订要求:每一小项放在一份文件保护袋中,再按照目录顺序要求放入三孔文件夹中,用索引纸分开(参考如下:齐心TC530AB 文件夹A4打孔夹背宽40mm 1.5寸3孔O型档案夹,蓝色天猫+%BF%D7+o%D0%CD%BC%D0+%C6%EB%D0%C4 。
3孔O型夹齐心蓝色)。
2、将电子档或扫描件发邮箱:.。