2_4_卤代苯甲酰基_苯胺的合成研究_戴立言
- 格式:pdf
- 大小:152.16 KB
- 文档页数:5


1,4-二(4-甲酰氯基苯甲酰基)苯的合成与表征祝志芳;钟鸣;易志群;余雯雯;宋琤;宋才生【摘要】A novel compound 1,4-di(4-methylbenzoyl)benzene (DMBC) was synthesized with terephthaloyl chloride (TPC) and toluene by Friedel-Crafts electrophilic aromatic substitution in the presence of anhydrous aluminum chloride as a catalyst. The yield of DMBB was 82%. Then 1,4-di(4-carboxyl benzoyl)benzene (DMBB) was obtained from DMBA by catalytic oxidation in the mixture of pyridine and water and potassium permanganate as oxidation agent. The yield of DMBA was 85%. 1,4-di(4-chloro formyl benzoyl)benzene DMBC was prepared by acidylated the DMBB with thionyl chloride as a acylating agent and N,N-dimethylformamide(DMF) as a catalyst. The yield of DMBC was 85%. The overall yield of the three-step reaction for DMBC was 59%. The chemical structure was characterized by FT-IR, 1H NMR and elemental analysis. The results showed that compound obtained as expected.%以甲苯、对苯二甲酰氯(TPC)为原料,无水三氯化铝为催化剂,通过亲电取代反应合成1,4-二(4-甲基苯甲酰基)苯(DMBB),产率为82%.在吡啶/水混合体系中用高锰酸钾将DMBB氧化得到1,4-二(4-甲羧基苯甲酰基)苯(DMBA),产率为85%.DMBA与二氯亚砜在DMF催化下反应合成1,4-二(4-甲酰氯苯甲酰基)苯(DMBC),产率为85%.3步总产率为59%.用FT-IR、1H NMR和元素分析等对化合物进行了表征.研究表明:化合物具有预期的结构和较高的纯度.【期刊名称】《江西师范大学学报(自然科学版)》【年(卷),期】2011(035)001【总页数】3页(P21-23)【关键词】1,4-二(4-甲酰氯苯甲酰基)苯;氧化;酰化;合成【作者】祝志芳;钟鸣;易志群;余雯雯;宋琤;宋才生【作者单位】江西师范大学化学化工学院,江西,南昌,330022;江西师范大学化学化工学院,江西,南昌,330022;江西师范大学化学化工学院,江西,南昌,330022;江西师范大学化学化工学院,江西,南昌,330022;江西师范大学化学化工学院,江西,南昌,330022;江西师范大学化学化工学院,江西,南昌,330022【正文语种】中文【中图分类】O623.62耐高温聚合物及其复合材料由于其优越的综合性能, 在航空、航天、核工业、微电子、机械工业等领域有广阔的应用前景. 目前使用最广的特种工程塑料有: 聚芳醚酮、聚苯硫醚、聚芳醚砜、聚芳酰胺、聚酰亚胺和聚芳酯等品种[1-3]. 制备高纯度的酰氯单体是合成高性能聚合物的关键之一, 如 Du Pont公司以间苯二甲酰氯(IPC)、对苯二甲酰氯(TPC)和二苯醚(DPE)共缩聚合成的聚芳醚酮酮(PEKK)[4-7],IPC与间苯二胺溶液缩聚制得的聚间苯二甲酰间苯二胺纤维(商品名Nomex)[8], TPC与对苯二胺经低温溶液缩聚合成聚对苯二甲酰对苯二胺(PPTA), 再经浓硫酸干喷湿纺制得的高强、高模芳纶1414纤维(Kevlar纤维)[9]. 本文从分子设计出发, 以甲苯、对苯二甲酰氯等为原料通过亲电取代、氧化、酰化等步骤, 制备了含多羰基结构的1, 4-二(4-甲酰氯苯甲酰基)苯. 作为一种结构新型的芳二酰氯单体可与芳二醚、芳二胺及芳二酚单体共缩聚制备一系列多羰基结构单元的聚芳醚酮、聚芳酰胺、聚芳酯等耐高温聚合物. DMBC的合成路线见Scheme 1.1.1 主要仪器和试剂Perkin-Elmer SP one FT-IR光谱仪(美国), KBr压片; Bruker Avance 400 MHz 型核磁共振仪(瑞士),溶剂为CDC13及氘代DMSO, TMS为内标; PE2400型元素分析仪; 毛细管法测定化合物的熔点(温度计未校正).甲苯, 天津市恒兴化学试剂制造有限公司, 重蒸后加0.4 nm分子筛干燥备用; 对苯二甲酰氯(TPC),工业品, 重蒸后熔点为 84~85 ℃, 南昌双菱化工厂;N,N-二甲基甲酰胺(DMF)、二氯乙烷(DCE)均为工业品, 重蒸后加入 0.4 nm分子筛干燥备用; 石油醚、高锰酸钾、吡啶(Py)、氯化亚砜、无水AlCl3、乙醇等为市售, 未经处理直接使用.1.2 合成方法1.2.1 1,4-二(4-甲基苯甲酰基)苯(DMBB)的合成在装有电动搅拌器及干燥装置的洁净 500 mL三颈瓶中, 加入 50.10 g无水 AlCl3, 60 mL甲苯(51.96 g), 搅拌并用冰盐浴冷却至-10 ℃左右, 缓慢滴加5 mL NMP, 分批加入30.50 g(0.15 mol)TPC,反应1 h后, 撤掉冰盐浴在20~30 ℃继续反应4~6 h.将反应混合液慢慢加入到有碎冰的约 150 mL蒸馏水中同时搅拌, 蒸馏回收过量甲苯. 析出的沉淀物经稀碱浸泡, 过滤, 水洗至中性, 粗产品用乙醇重结晶2~3次, 得到白色晶体38.40 g, 产率为82%(以TPC计), m.p.: 182~183 ℃.1.2.2 1,4-二(4-羧基苯甲酰基)苯(DMBA)的合成在装有回流冷凝管和电动搅拌器的1 000 mL三颈瓶中, 加入15.70 g (0.05 mol) 1,4-二(4-甲基苯甲酰基)苯(DMBB)、100 mL吡啶、90 mL蒸馏水, 搅拌下加热至90~100 ℃, 分批加入63.2 g(0.4 mol)高锰酸钾.反应8~9 h后, 趁热过滤, 用热水洗涤滤饼数次. 滤液蒸馏回收吡啶和水的恒沸物, 残留液经盐酸酸化析出白色固体, 过滤. 粗产品经碱溶、酸析、过滤, 干燥后得到白色粉末15.80 g. 产率为85%(以TPC计).1.2.3 1,4-二(4-甲酰氯苯甲酰基)苯(DMBC)的合成在装有回流冷凝管的干燥洁净 500 mL三颈瓶中, 加入 18.70 g(0.05 mol)1,4-二(4-羧基苯甲酰基)苯和140 mL氯化亚砜, 并加入少量DMF或Py作催化剂, 加热回流, 反应至溶液澄清. 蒸馏回收过量的氯化亚砜, 用体积比为1∶1的石油醚和DCE重结晶 2次, 过滤. 干燥后得到白色晶体 17.60 g. 产率为85% (以TPC计), m.p. : 204~205 ℃.2.1 1,4-二(4-甲基苯甲酰基)苯(DMBB)的红外和核磁共振图谱分析FT-IR,v/cm-1: 1 650.92(羰基); 1 605.59, 1 560.17(苯环骨架); 1 382.13(甲基).1H NMR (CDCl3),δ: 2.459(s,6H,Ha), 7.302(m,4H,Hb), 7.765(m,4H,Hc), 7.859(m,4H,Hd), 如图1所示. 元素分析(C22H18O2)实测值(计算值)/%: C,84.026(84.051); H, 5.781(5.771).2.2 1,4-二(4-羧基苯甲酰基)苯(DMBA)的红外和核磁共振图谱分析FT-IR,v/cm-1: 3 438.70(羧羟基); 1 703.59, 1 649.92(芳酮, 羰基); 1 608.89, 1 501.02(苯环骨架).1H NMR(DMSO-d6,),δ: 13.108(s,2H,Ha), 8.106(m,4H,Hb), 7.915(m,8H, Hc, d), 如图 2所示. 元素分析(C22H14O6)实测值(计算值)/%:C,70.562(70.588); H,3.776(3.769).2.3 1,4-二(4-甲酰氯苯甲酰基)苯(DMBC)的红外和核磁共振图谱分析FT-IR,v/cm-1: 1 767.23, 1 656.81(羰基);1 606.79, 1498.05(苯环骨架);756.33(酰氯碳氯).1H NMR (CDCl3)如图 3 所示.δ:8.263 (m,4H,Ha),7.951(m,8H, Hb,c). 元素分析(C22H12O4Cl2)实测值(计算值)/%: C, 64.226(64.257);H,2.953(2.941).以甲苯和对苯二甲酰氯(TPC)为原料经傅-克亲电取代、氧化、酰化3步反应合成了一种结构新型的芳二酰氯1,4-二(4-甲酰氯苯甲酰基)苯, 经FT-IR、1H NMR和元素分析表明: 目标产物具有预期的结构和较高的纯度, 可作为一种新型改性单体,制备主链含多羰基的聚芳醚酮、聚芳酯和及聚芳酰胺等耐高温聚合物.【相关文献】[1] 张云鹤, 王冬, 牛亚明, 等. 新型含萘含氟聚芳醚酮的合成与表征[J]. 高等学校化学学报, 2005, 26(7): 1378-1380.[2] 吴忠文. 特种工程塑料聚醚砜、聚醚醚酮树脂国内外研究、开发、生产现状[J]. 化工新型材料, 2002, 30(6): 15-18.[3] 周其凤, 范星河, 谢晓峰. 耐高温聚合物及其复合材料——合成、应用与进展[M]. 北京: 化学工业出版社, 2004:113-134.[4] 唐旭东, 王艳, 贺征华. 特种工程塑料聚芳醚酮的研究进展[J]. 天津科技大学学报, 2006,21(1):29-32.[5] 钟鸣, 宋才生, 童永芬, 等. 含间苯基及甲基侧基聚芳醚砜醚酮酮的合成与表征[J]. 江西师范大学学报: 自然科学版, 2005, 29(3): 193-195.[6] 付长清, 宋才生, 刘勇军, 等. 聚芳醚酮酮、聚芳醚醚酮酮无规共聚物的合成与性能研究[J]. 江西师范大学学报: 自然科学版, 2005, 29(5): 377-380.[7] Wen Hong-li, Song Cai-sheng, Tong Yong-fen, et al.Synthesis and properties ofpoly(aryl ether sulfene ether ketone ketone) (PESEKK)[J]. Journal of Applied Polymer Science, 2005, 96: 489-493.[8] 裴晓辉. 新型半芳香聚酰胺的合成与表征[D]. 河南: 郑州大学, 2005.[9] 杨拯, 潘婉莲, 曹煜彤, 等. 芳纶 1414的聚合研究[J]. 材料导报: 网络版, 2006(3): 10-13.。


哌嗪衍生物合成工艺进展田忠社;韦雄雄;梁建国;刘荣杰;张彦【摘要】哌嗪衍生物在医药和化工领域有广泛的应用,详细叙述了烷基类哌嗪衍生物、苯基类哌嗪衍生物、二酮类哌嗪衍生物、异黄酮类哌嗪衍生物、雌激素类哌嗪衍生物的合成方法,并指出了哌嗪衍生物合成过程存在的问题和以后的研究方向.%Piperazine derivatives in the pharmaceutical and chemical industry have a wide range of applications. The synthesis method of species of alkyl piperazine derivatives, phenyl piperazine derivatives, diketopiperazines derivatives, isoflavones-piperazine derivatives, estrogen piperazine derivatives were described and the problems of the synthesis process of piperazine derivatives and research directions were pointed out.【期刊名称】《应用化工》【年(卷),期】2011(040)009【总页数】5页(P1648-1652)【关键词】哌嗪衍生物;工艺;进展【作者】田忠社;韦雄雄;梁建国;刘荣杰;张彦【作者单位】西北大学化工学院,陕西西安 710069;西北大学化工学院,陕西西安710069;西北大学化工学院,陕西西安 710069;西北大学化工学院,陕西西安710069;西北大学化工学院,陕西西安 710069【正文语种】中文【中图分类】TQ254;R914.5;O613.52哌嗪(环胺杂环化合物)是一种重要的有机化工中间体,主要应用于医药、农药、染料等行业,哌嗪衍生物同样广泛应用于医药和化工领域,其中重要的有5类:异黄酮类、烷基类、雌激素类、二酮类和苯基类[1-3],哌嗪衍生物合成方法的研究是目前面临的重要课题。
![一种苯溴马隆的制备方法[发明专利]](https://img.taocdn.com/s1/m/5ba7eee952ea551811a687a4.png)
专利名称:一种苯溴马隆的制备方法
专利类型:发明专利
发明人:黄丽荣,胡铁军,李想,白跃飞,宋海生申请号:CN201210118521.8
申请日:20120420
公开号:CN102659727A
公开日:
20120912
专利内容由知识产权出版社提供
摘要:一种应用于药物合成领域中的苯溴马隆的制备方法,该方法包括下列步骤:以2-乙基苯并呋喃和对甲氧基苯甲酰氯为起始原料,在催化剂的参与作用下进行傅克酰基化反应,制得2-乙基-3-对甲氧基苯甲酰-苯并呋喃;所得2-乙基-3-对甲氧基苯甲酰-苯并呋喃与盐酸吡啶进行去甲基化反应,使用甲苯带水的方法去除了反应体系中的水分,制得2-乙基-3-对羟基苯甲酰-苯并呋喃;所得2-乙基-3-对羟基苯甲酰-苯并呋喃与溴化物进行溴代反应,制得苯溴马隆;2-乙基苯并呋喃和对甲氧基苯甲酰氯充分反应后用盐酸酸解,萃取得到2-乙基-3-对甲氧基苯甲酰-苯并呋喃。
该发明在傅克酰基化反应中,使用二氯甲烷、氯仿等卤代烃替换二硫化碳,简化后处理过程;在溴代反应中,将腐蚀性强、对人体危害大、污染环境的溴素改换成溴化物。
申请人:东北制药(沈阳)科技发展有限公司
地址:110027 辽宁省沈阳市沈阳经济技术开发区昆明湖街8号
国籍:CN
代理机构:沈阳维特专利商标事务所(普通合伙)
代理人:甄玉荃
更多信息请下载全文后查看。
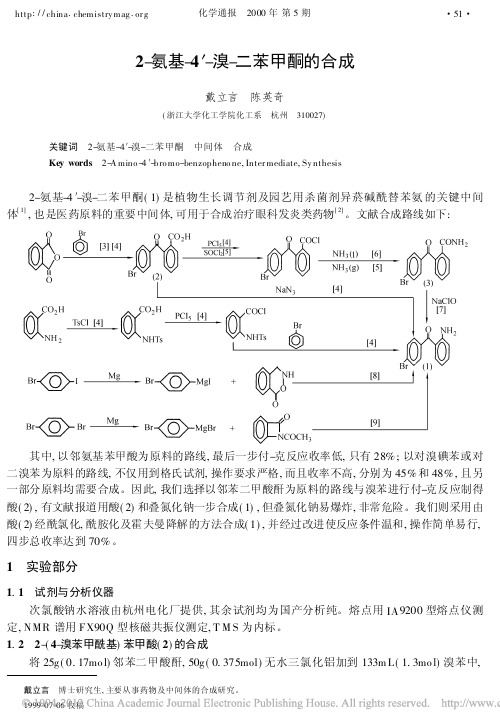
2-氨基-4′-溴-二苯甲酮的合成戴立言 陈英奇(浙江大学化工学院化工系 杭州 310027)戴立言 博士研究生,主要从事药物及中间体的合成研究。
1999-07-06收稿关键词 2-氨基-4′-溴-二苯甲酮 中间体 合成Key words 2-A mino -4′-bro mo-benzopheno ne,Inter mediate,Sy nthesis2-氨基-4′-溴-二苯甲酮(1)是植物生长调节剂及园艺用杀菌剂异菸碱酰替苯氨的关键中间体[1],也是医药原料的重要中间体,可用于合成治疗眼科发炎类药物[2]。
文献合成路线如下:其中,以邻氨基苯甲酸为原料的路线,最后一步付-克反应收率低,只有28%;以对溴碘苯或对二溴苯为原料的路线,不仅用到格氏试剂,操作要求严格,而且收率不高,分别为45%和48%,且另一部分原料均需要合成。
因此,我们选择以邻苯二甲酸酐为原料的路线与溴苯进行付-克反应制得酸(2),有文献报道用酸(2)和叠氮化钠一步合成(1),但叠氮化钠易爆炸,非常危险。
我们则采用由酸(2)经酰氯化,酰胺化及霍夫曼降解的方法合成(1),并经过改进使反应条件温和,操作简单易行,四步总收率达到70%。
1 实验部分1.1 试剂与分析仪器次氯酸钠水溶液由杭州电化厂提供,其余试剂均为国产分析纯。
熔点用IA 9200型熔点仪测定,NMR 谱用FX90Q 型核磁共振仪测定,T M S 为内标。
1.2 2-(4-溴苯甲酰基)苯甲酸(2)的合成将25g (0.17mo l)邻苯二甲酸酐,50g (0.375mol)无水三氯化铝加到133m L(1.3mo l)溴苯中,・51・http ://china .chemistry mag .or g 化学通报 2000年第5期反应立即剧烈进行,并放出氯化氢气体。
自然升温至50℃,当氯化氢气体放出速度减慢时,加热并保温在50℃。
继续反应5h ,冷却至室温,在搅拌下缓慢倒入冰水中,然后加入饱和碳酸钠溶液调节pH 至12,水汽蒸馏回收溴苯(可得110mL,169.4g ,1.1mol),残液用稀硫酸调节pH 至1,产生大量的白色沉淀,抽滤,烘干,粗品用120mL 无水乙醇重结晶,得到2-(4-溴苯甲酰基)苯甲酸(2)45.5g 。
2004 年2 月 Journal of Chemical Engineering of Chinese Universities Feb. 2004 文章编号:1003-9015(2004)01-0094-052-(4-卤代苯甲酰基)苯胺的合成研究戴立言, 吴彩娟, 陈英奇, 吴兆立(浙江大学材料与化工学院,浙江杭州,310027)摘要:研究了重要的有机中间体2-(4-卤代苯甲酰基)苯胺的通用合成路线,是以邻苯二甲酸酐为原料,与卤代苯(分别为氟苯、氯苯和溴苯)进行付-克反应得到羧酸;羧酸经酰氯化、酰胺化,制得邻苯甲酰基苯甲酰胺衍生物,然后再经过霍夫曼降解合成了标题化合物。
对主要步骤反应条件进行了优化,得出比较合理的条件为:付-克反应,催化剂:AlCl3,配料比:1:2.2:6.4(苯酐:AlCl3:卤代苯);同时使用卤代苯既作反应试剂,又作溶剂,未反应的卤代苯回收套用,使收率基本定量;酰胺化反应氨气流量:10mL⋅min−1,使反应得以室温进行,无需制冷;四步反应总收率可达70%。
关键词:2-(4-卤代苯甲酰基)苯胺; 2-(4-氟苯甲酰基)苯胺; 2-(4-氯苯甲酰基)苯胺; 2-(4-溴苯甲酰基)苯胺;中间体; 合成中图分类号:O621.3; TQ246.31 文献标识码:A1 前言2-(4-卤代苯甲酰基)苯胺(1)(X=F、Cl、Br)是重要的有机原料和医药中间体,可用于合成植物生长调节剂和园艺杀菌剂异菸碱酰替苯胺[1]和还原酶HMG-CoA的阻聚剂NK-104[2],也用于合成2-氨基-3-卤代苯甲酰基苯乙酰胺衍生物,用于治疗眼科发炎症状[3],还可用于合成4-卤代苯-2-哌嗪喹啉衍生物,目前又发现该类物质具有抗溃疡作用[4]。
其主要合成路线如下:收稿日期:2001-12-20; 修订日期:2002-05-30。
作者简介:戴立言(1971-),男,辽宁开原人,浙江大学副教授、博士。
通讯联系人:戴立言,E-mail: dailiyan@路线(ⅰ):X=F、Br该路线有文献报道,是以邻苯二甲酸酐为原料,与卤代苯在无水三氯化铝催化下进行付-克反应制得羧酸(2),羧酸再经过酰氯化、酰胺化,得酰胺(3),然后经过霍夫曼降解得到标题化合物(1),收率为65%。
研究论文依托度酸的合成工艺戴立言! 王晓钟! 陈英奇(浙江大学材料与化工学院制药工程研究所9 浙江 杭州 310027)摘要C 对非甾体抗炎药依托度酸的合成工艺进行了 研 究9 以 生 产 氯 霉 素 原 料 对 硝 基 乙 苯 时 产 生 的 大 量 副 产 物邻硝基乙苯为原料9 先还原得邻乙基苯胺9 然后经重 氮 化\ 亚 硫 酸 钠 还 原 得 邻 乙 基 苯 肼 盐 酸 盐9 再 与 293-二 氢 呋 喃反应得7-乙基色醇9 不经分离9 直接与3-氧代戊酸甲酯 缩 合 成 环\ 水 解 制 得 标 题 化 合 物. 各 步 反 应 的 最 佳 反 应条件 (反应温度9 反应时间9 摩尔收率) 分别为9 硝基还原C 回流93h 996.2 ; 重氮化C 0 90.5h ; 重 氮盐还原C 70~75 93h 9(两步收率)92.5 ; 合成7-乙基色醇C 回流93h ; 制备依托度酸甲酯C 0 91.5 h 9(两步)63.0 ; 酯水解C 回流92.5h 995.0 . 所得依托度酸甲酯和依托度酸物性与文献报道一致.关键词C 依托度酸; 药物合成; 非甾体抗炎药 中图分类号C 621.3 文献标识码C A 文章编号C 0438-1157 (2005)08-1536-05S yn t h e s i s o f e t o d o l a c D A I L i y a n !W A N G X i a o z h o n 9!C H E N Y i n 90i (I n s t i t u t e O f P h a r m a c e u t i c a l E n g i n e e r i n g 9C O l l e g e O f M a t e r i a l S c i e n c e a n d C h e m i c a l E n g i n e e r i n g 9Z h e j i a n g U n i u e r s i t $9 a n g z h O u3100279Z h e j i a n g 9Ch i n a ) A b s t r a c t C S y n t h e s i s O f e t O d O l a c 9an O n -s t e r O i d a l a n t i -i n f l a m m a t O r y a g e n t 9W a ss t u d i e d .I n t h e s t u d y O - n i t r O e t h y l b e n z e n ea s r a W m a t e r i a l W a s r e d u c e db y F e /h y d r O c h l O r i c a c i d .T h e i n t e r m e d i a t e O -e t h yl a n i l i n e c O u l db ed i a z O t i z e da n d r e d u c e db y s O d i u m s u l f i t e t O g i v e O -e t h y l p h e n y l h y d r a z i n e 9a n d7-e t h y l t r y p t O p h O l W a s O b t a i n e d b y c O n d e n s a t i O n O f O -e t h y l p h e n y l h y d r a z i n ea n d293-d i h y d r O f u r a n i n i s O b u t a n O l a ss O l v e n t . F i n a l l y 7-e t h y l t r y p t O p h O l W i t h O u t s e p a r a t i O n r e a c t e d W i t h m e t h y l 3-O X O p e n t a n O a t ea n d W a s h y d r O l yz e d t O g i v e t h e t i t l e c O m p O u n d .T h e O p t i m a l r e a c t i O n c O n d i t i O n s (t e m p e r a t u r e 9t i m e 9 m O l a r yi e l d )W e r e a s f O l l O W s .R e d u c t i O n O f n i t r O g r O u p C re f l u X 93 h 996.2 ;d i a z O t i z a t i O n C 0 90.5 h ;r e d u c t i O n O f d i a z O n i u m s a l t C 70_75 93 h 992.5 (t W O s t e p s );s y n t h e s i s O f 7-e t h y l t r y p t O ph O l C r e f l u X 93 h ; p r e p a r a t i O n O f e t O d O l a c m e t h y l e s t e r C 0 91.5 h 963.0 (t W O s t e p s );h y d r O l y s i s C r e f l u X 92.5 h 9 95.0 .T h e s t r u c t u r e s O f e t O d O l a c a n d i t s m e t h y l e s t e r W e r e t h e s a m ea s t h O s e r e pO r t e d i n l i t e r a t u r e .K e y Wo r d s C e t O d O l a c ;p h a r m a c e u t i c a l s y n t h e s i s ;n O n -s t e r O i d a l a n t i -i n f l a m m a t O r y d r u g a n t i -i n f l a m m a t O r y d r u g s 9N S A I D s )9 镇 痛 抗 炎 作 用强9 临床上广泛用于术后疼痛的治疗9 缓解类风湿性关节炎和骨关节炎的症状9 延缓关节炎所引起的骨病理改变[1].文献报道[293]依托度酸的合成主要是由7-乙基 引 言依托 度 酸 (e t O d O l a c 9 商 品 名 U l t r a d O l 9L O d - i n e ) (1) 是20 世纪70 年代中期由 ~u m b e r 研究 开发的吡喃糖羧 酸 类 非 甾 体 抗 炎 药 (n O n -s t e r O i d a l 2004-06-28 收到初稿92004-08-16 收到修改稿.联系人及第一作者" 戴立言 (1971_ )9 男9 博士9 副教授. R e c e i v e dd a t e "2004-06-28.C o r r e s P o n d i n 9au t h o r " D r . D A I L i y a n .E - m a i l "d a i l i y a n @ 妥、妥第8期 戴立言等= 依托度酸的合成工艺- 1537- 色醇 <4> 与3-氧代戊酸甲酯或3-甲氧基-2-戊烯酸甲酯反应得依托度酸甲酯 然后水解即可制得. 其中关键之 处 在 于 7-乙 基 色 醇 的 合 成 文 献 报 道 有 两种合成方法 方法一 4 5]以工业生产氯霉素中间 体对硝基 乙 苯 的 副 产 物 邻 硝 基 乙 苯 为 原 料 经 锡 粉~ 盐酸还原得邻乙基苯胺 然后与水合氯醛~ 盐 酸羟胺在酸性条件下形成肟基乙酰胺衍生物 再在 浓硫酸中 环 合 得 7-乙 基 吲 哚 二 酮 用 氢 化 铝 锂 还 原得 7-乙 基 吲 哚 与 草 酰 氯 反 应 再 经 酯 化~ 硼 氢化钠还原制得; 方法二 3 6~9]也是以邻硝基乙苯 为原料 经硫化钠或铁粉还原得邻乙基苯胺 然后 经重氮化后 用亚硫酸钠~ 亚硫酸氢钠或氯化亚锡 还原得邻乙 基 苯 肼 盐 酸 盐 再 与 2 3-二 氢 呋 喃 在 1 4-二氧六环中回流反应制得. 分析上述合成方法 方法一不仅路线长 而且 原料以及所用的还原剂价格昂贵 不适宜工业化生 产; 本文针对方法二进行了重点改进研究 以工业 生产氯霉素中间体对硝基乙苯时产生的大量廉价副 产物邻硝基乙苯为原料 通过调节铁粉的用量 高 收率获得了还原产物邻乙基苯胺. 在制备邻乙基苯 肼盐酸盐的过程中 按照文献 6 8] 的方法 还 原重氮盐时将预制好的低温重氮盐溶液倒入 85~ 90的亚 硫 酸 钠 溶 液 中 按 此 方 法 进 行 实 验 收 率极低 而且产生大量黑色~ 有刺鼻气味的焦油状物质; 而文献 9] 使 用 氯 化 亚 锡 还 原 重 氮 盐 成本高. 针对于此 作者分析 在还原重氮盐的实验 过程中不能将预制好的盐酸重氮盐直接倒入热的亚 硫酸钠溶液中去 而是在低温时将其加到冷的亚硫 酸钠溶液中 可以避免重氮盐受热分解 保温一段 时间后 将盐酸重氮盐转化为亚硫酸重氮盐 再缓 慢升温 还 原 效 果 非 常 好 产 品 为 类 白 色 片 状 晶 体 收率高达92.5 . 另外 合成7-乙基色醇时 文献 7] 使 用 价 格 较 高 的 1 4-二 氧 六 环 为 溶 剂 反应完毕 用 硅 胶 柱 层 析 法 获 得 较 纯 的 7-乙 基 色 醇 再与3-氧代 戊 酸 甲 酯 或 3-甲 氧 基-2-戊 烯 酸 甲 酯反应得依托度酸甲酯 不仅增加了分离步骤 而 且分离收率低 损失较大. 本实验改用廉价的异丁 醇为溶剂 与 2 3-二 氢 呋 喃 反 应 完 毕 回 收 溶 剂 后 不经分离提纯 直接在甲苯和少量乙醇中以浓 硫酸为缩合 剂 与 3-氧 代 戊 酸 甲 酯 反 应 得 依 托 度 酸 甲酯 反应完毕分层 得到的甲酯物溶解在甲苯中 与浓硫酸分离~ 脱色 浓缩即可得纯的甲酯物 杂 质在浓硫 酸 和 乙 醇 层 不 仅 克 服 了 7-乙 基 色 醇 难 以分离提 纯 的 困 难 避 免 了 7-乙 基 色 醇 纯 化 时 的 损失 而且简化了反应操作 同时浓硫酸层经溶剂 萃取除去杂质后可以回收套用 减少了环境污染 收率较高.具体合成路线如下=己烷及甲醇等均为市售分析纯.实验部分1 分析仪器及型号E l e c t r O t h e r m a l I A 9200 型 熔 点 仪 A g i l e n t 6820型气相色谱仪. 1.2 主要试剂及规格邻硝基乙苯 <96 浙江神鹰医药化工有限公 司> 亚硝酸钠 <99浙江上虞三和医药化工有 限公司> 2 3-二 氢 呋 喃 <99江 苏 省 武 进 市 有 机化工厂> 异丁醇 <99.5浙江华义医 药 有 限 公司> 还 原 铁 粉~ 浓 盐 酸~ 浓 硫 酸~ 亚 硫 酸 钠~ 无水硫酸钠~ 溶剂甲苯~ 乙醇~ 二 氯 甲 烷~ 氯 仿~ 1.1 典型实验 1.3 1.3.1 邻乙基苯胺 !2" 向500 m l 三口瓶中加入 铁粉 <89g 1.59 m O l > 将 水 <105g > 与 浓 盐 酸<8 m l >混 合 缓 慢 加 到 铁 粉 中 升 温 至 100化 工 学 报 第56卷 ~ 1538 ~ 滴加完毕 回 流 反 应 3h .反 应 完 毕 水 汽 蒸 馏 收集馏出液 分出上层油层为71 m l得黄色透明 反应完毕 静置分层 分出上层甲苯层 下层硫酸再用40 m l >3 甲苯萃 取. 合 并 甲 苯 层 倒 入 5 碳酸氢钠溶液中 分出有机层 用无水硫酸钠 干燥 浓缩 回 收 溶 剂 至 约 剩 下 20 m l 甲 苯 加 入20 m l 石油 醚 冷 冻 抽 滤 用 少 量 石 油 醚 淋 洗 干燥 得 白 色 固 体 27.5 g 收 率:63.0 m p : 液 体 g 气 相 色 谱 含 量 99.5 收 77.1 率:96.2 .邻乙基苯肼盐酸盐向 250 m l 三 口 !3" 1.3.2 瓶中加入浓盐酸 <53 m l > 和水 <50 m l> 降温至0 缓慢滴加邻乙基苯胺 <24.2g 0.2 m O l > 滴 加过程 中 控 制 温 度 不 超 过 0 约 0.5h 滴 加 完 毕 保温在0 剧烈搅拌下 缓慢滴加预先配制 好的亚硝酸钠水溶液 <14.5g 0.21 m O l 亚硝酸钠 溶于30 m l 水>. 滴加开始时 反应 混 合 物 比 较 黏 稠 颜色逐渐变成粉红色 随着滴加的进行 逐渐变稀至全溶. 滴加完毕 继续保温搅拌 0.5h . 与 此同时 向另一1000 m l 三口瓶中加入固体亚硫酸 钠 <63g 0.5 m O l > 再加入350 m l 水 降温至0 加入25 m l 浓 盐 酸 再 加 入 25g 冰 然后 将做好的重氮盐溶液快速倒入此亚硫酸钠溶液中 在0 保温30 m i n 撤去冰浴 自然升至室温再保温 30 m i n此时有大量固体析出. 保温毕 缓慢加热 至50 时 固体全溶 继续加热至70~75 保温反应3h 反应混合物变成浅红色透明溶液. 然后向此溶液中缓慢加入75 m l 浓盐酸此时有大量二 氧化硫气 体 放 出. 加 毕 于 70 搅 拌 30 m i n 降 温至50 即有 大 量 片 状 晶 体 析 出 继 续 降 温 至 0析出 固 体 抽 滤. 滤 饼 用 少 量 18 盐酸 漂 洗 烘干 用乙醇/水混合溶剂重结晶得类白色片状晶130~131 与文献 I 10I 一致. 依托度酸 !1" 将9.6g 依托度酸甲酯加 1.3.4 到150 m l 甲醇中 再加入25 m l 30 的液碱加热回流 约2.5h 反应完毕 液相色谱检测反应终 点. 反应 完 毕 减 压 回 收 溶 剂 向 剩 余 物 中 加 入100m l 水 用40m l 二氯甲烷萃取除杂质 分去有 机层 水层用 18 盐酸调节 p~ 至 4 析出 固 体 抽滤干燥 用氯仿/己烷重结晶得白色固体 8.7g含量98.9 收率:95.0 m p :146.2~147 <文献 I 3I m p :146.1~146.8 文献 I7I m p : 146~147 文献 I 10I m p :145~147 >.结果与讨论2 铁粉用量对邻硝基乙苯还原反应的影响 理论上 铁粉与相应被还原的硝基化物的摩尔比为2.25:1 通常还要加入少量盐酸 使部分铁 粉生成氯化亚铁 形成电解质溶液 增大了电解质2.1 的传导速 率 加 速 了 铁 氧 化 成 的 速 度 从 F e 3O 4 而加快 硝 基 化 合 物 的 还 原. 但 实 验 过 程 中 发 现铁粉用量对还原 收 率 有 一 定 的 影 响 本 文 对 还 原邻硝基乙苯的铁 粉 最 佳 用 量 进 行 了 研 究 具 体 见 表1.依托度酸甲酯 !5" 向 500 m l 烧 瓶 中 加 1.3.3入邻乙基苯肼盐酸盐 25g <0.145 m O l > 5 m l 水300 m l 异丁醇 缓 慢 滴 加 2 3-二 氢 呋 喃 <11.2g12.1 m l 0.16 m O l > 约 30 m i n 滴加 完 毕 加 热 回流 反应3h 薄层分析检测反应终点 展开剂 为: 丙酮/氯仿 <体 积 比 1:4> 其 中 比 移 值 R f 为0.95 的是原料苯肼 R f 为0.5 的是产物色醇. 反应完毕 减压蒸馏回收溶剂至干 溶剂回收率可 达95 以上 剩 余 物 为 粗 品 7-乙 基 色 醇 以 及 反 应 生成的无机盐氯化铵. 向上述粗品中加入乙 T a b l e l E f f e c t o f i r o na m o u n t o n r e d u c i n gr e a c t i o no f -n i t r o e t h yl b e n z e n e I r O np O W d e r /g m O l I r O n :O -n i t r O e t h y lb e n z e n e <m O l> Y i e l d/ 83.4 1.490 85.3 1.523 87.2 1.556 89.0 1.589 93.0 1.6562.25:12.30:1 2.35:1 2.40:1 2.50:170.7 82.3 93.5 96.2 96.3N O t e :O -n i t r O e t h y l b e n z e n e 100g <0.66 m O l > r e f l u X i n3h .实验证明 铁粉与邻硝基乙苯摩尔比为2.4:1时比较 理 想 铁 粉 用 量 再 增 加 收 率 提 高 已 不 明 显 而 铁 粉 用 量 减 少 反 应 不 完 全 导 致 收 率降低.醇 200 m l 甲 苯 3-氧 代 戊 酸 甲 酯 <21g 0.159 m O l > 冰盐水降温至 0 保温缓慢滴加 50g浓 硫酸 约 1h 滴 毕 薄 层 分 析 检 测 反 应 终 点 约 1.5h 反应完毕.R f 为0.5 的是色醇 R f 为0.95第8期 戴立言等I 依托度酸的合成工艺1539亚硫酸钠用量对邻乙基苯肼盐酸盐收率的 影响用亚硫酸钠还原重氮盐时9 首先重氮盐与亚硫 酸钠形成偶氮亚硫酸钠9 再与一分子亚硫酸钠形成 氢化偶氮二亚硫酸钠9 然后加热在酸性条件下水解 即得苯肼盐酸盐9 因此理论上亚硫酸钠与重氮盐的 摩尔比为2:19 但 实 际 实 验 过 程 中9 适 当 增 加 亚 硫酸钠的用量9 可以提高反应的收率. 本文对其最 佳用量比进行了研究9 具体见表2.剂回流温度下反应收率最高.在此9 又 选 用 了 194-二 氧 六 环 甲 苯 氯 苯 和异丁醇等 沸 点 相 近 但 极 性 不 同 的 溶 剂 进 行 了 研 究9 结果 表 明 以 极 性 最 大 的 异 丁 醇 为 溶 剂9 收 率 最高.2.2 E f f e c t o f 2"3-d i h y d r o f u r a na m o u n t o n y i e l do f 7-e t h y l t r y p t o ph o l T a b l e 4 293-d i h y d r O f u r a n g 9m l 9m O l293-d i h y d r O f u r a n I h y d r a z i n ec O m p O u nd (m O l) Y i e l d10.2911.090.14510.7911.690.15211.2912.190.16011.7912.790.167E f f e c t o f s o d i u m s u l f i t e !s a m o u n t o n y i e l d1.00:11.05:1 1.10:11.15:157.5 61.4 63.063.3T a b l e 2 o f -e t h y l p h e n y l h y d r a z i n eh yd r o c h l o r i de S O d i u m s u lf i t eg 9m O l S O d i u m s u l f i t e :a m i n Oc O m p O u nd (m O l) Y i e l d12.2913.390.1741.20:162.650.490.40 55.490.4460.590.4863.090.5065.590.522.0:12.2:1 2.4:1 2.5:1 2.6:181.084.3 87.8 92.5 92.2N O t e I O -e t h y l p h e n y l h y d r a z i n eh y d r O c h l O r i d e 25g (0.145m O l)9 m e t h y l 3-O X O -p e n t a n O a t e21g (0.159 m O l )9i s O b u t a n O l a ss O l v e n t a n d r e f l u X .由表 可以看出9 9 二氢呋喃用量对收率的 4 2 3- 影响不是很明显9 主要是因为反应过程中9293-二氢呋喃首 先 与 水 反 应 生 成 中 间 体 4-羟 基 丁 醛9 此 过程非常 快9 然 后 4-羟 基 丁 醛 与 肼 形 成 腙 的 过 程 中又脱除一分子水9 因此9 在反应过程中9293-二 氢呋喃基本可以完全参加反应9 损失很小.N O t e I O -e t h y l a n i l i n e 24.2g (0.2 m O l )9s O d i u m n i t r i t e14.5g(0.21 m O l ).由表2 可以看出9 最佳的亚硫酸钠与邻乙基苯胺摩尔比为 2.5:19 用 量 继 续 增 加9 理 论 上 收 率 不应 该 降 低9 此 处 的 偏 差 应 该 是 由 实 验 误 差 引 起的.结 论3 反应温度 293-二氢呋喃用量以及溶剂种类 对7-乙基色醇收率的影响 合 成 依 托 度 酸 的 难 点 在 于 7-乙 基 色 醇 的 分 离 和纯化导致的收率低9 在此9 研究了反应温度 所 用反应溶剂 以 及 293-二 氢 呋 喃 用 量 对 其 收 率 的 影 响9 由于本 工 艺 采 用 不 分 离 出 7-乙 基 色 醇9 直 接 合成依托度酸甲酯的方法9 所以其收率以依托度酸 甲酯的产量为基准. 具体见表3 和表4.从表3 可以看出9F i s h e r 吲哚环的合成法在2.3 (1)以生产氯霉素原料对硝基乙苯时产生的大量副产物 邻 硝 基 乙 苯 为 原 料9 先 还 原 得 邻 乙 基 苯胺9 然后经重氮化 还原得邻乙基苯肼盐酸盐9 再与293-二 氢 呋 喃 反 应 得 7-乙 基 色 醇9 最 后 与 3-氧 代戊酸甲酯缩合成环 水解制得依托度酸9 总收率 为53.3 9 本工艺具有反应条件温和 操作简单 成本低等优点9 具有较好的工业化应用前景. (2) 用铁粉还原邻硝基乙苯9 铁粉与邻硝基乙 苯摩尔比为2.4:1. (3) 用亚硫酸钠还原邻乙基苯胺重氮盐9 最佳 的亚硫酸钠与邻乙基苯胺摩尔比为2.5:1.(4) 合成 7-乙 基 色 醇 时9 使 用 廉 价 的 异 丁 醇 为溶剂9 反 应 在 回 流 温 度 下 进 行9 最 佳 的 293-二 氢呋喃与邻乙基苯肼盐酸盐的摩尔比为1.1:1.T a b l e 3 E f f e c t o f r e a c t i o n t e m pe r a t u r e o n y i e l do f 7-e t h y l t r y p t o ph o lR e a c t i O n t e m p e r a t u r eY i e l d60 6570 7580 85 90 95r e f l u X26.439.2 47.7 58.263.0R e f e r e n c e s1 S h i J i n m i n (石 劲 敏 )9 L i D u a n (李 端 ). A c h i r a ln O n s t e r O i d a l a n t i -i n f l a mm a t O r y d r u g Of e t O d O l a c .C h i n e s e J O u r n a l O f C l i n i c a l P h a r m a c $ (中 国 临 床 药 学 杂 志 )9 N O t e I O -e t h y l p h e n y l h y d r a z i n eh y d r O c h l O r i d e 25g (0.145m O l)9 293-d i h y d r O f u r a n11.2g (12.1 m l 90.16 m O l )9m e t h y l 3-O X O -pe n - t a n O a t e21g (0.159 m O l )9i s O b u t a n O l a ss O l v e n t .化 工 学 报 第56卷 1540 O f P h a r m a c e u t i c a l s <中 国 医 药 工 业 杂 志 > 1998 2 <20>:89Pl a i n s b O r O C A D ~u m b e r L G . U S 4585877.1986 Ch e n Y O n g i e <陈 永 杰 > X u D a u n <徐 大 军 > ~a O J i n g <郝 晶 >.S y n t h e s i s O f O -e t h y l p h e n y l h y d r a z i n e W i t h s O d i u m s u l f i t e r e d u c t i O n . J O u r n a l O f S h e n $a n g I n s t i t u t e O f C h e m i c a l T e c h n O l O g $ <沈 阳 化 工 学 院 学 报 > 1999 13<4>:243C h e n Y O n g i e <陈 永 杰 > C h e n G u a n gX i n <陈 广 新 > Y i n ~O n g m e i <尹 红 梅 >.S y n t h e s i s O f O -e t h y l p h e n y l h yd r a z i ne . F i n e C h e m i c a l E n g i n e e r i n g <精细化工> 1996 13:28 C i t t e r i O A F a n c e l l i D P e s c eL . E P 302541.19892000 <5>:321L u r a gO D E V .U S 6066741.2000 Zh a n g Z h i l i u <张治柳> N i J u n <倪 俊> W a n g Y u r e n <王 愉 人 >. E X p e n r i m e n t a l r e s e a r c h f O r s yn t h e t i c p r O c e s s O f e t O d O l a c .C h i n a P h a r m a c e u t i c a l s <中 国 药 业 > 2001 1 <10>:3 ~a v a n t G O W . U S 4062869.1977N i P e i z h O u <倪沛洲> S h i X i n z h O n g <施欣忠> C h e n J iu n <陈 继 俊 >. S y n t h e s i s O f 7-e t h y l -1~-i n d O l e . J i a n gs u C h e m i c a l E n g i n e e r i n g <江苏化工> 1993 21 <1>:17 P a n F u y O u <潘 富 友> Y a n g J i a n g u O <杨 建 国>.S yn t h e s i s Of 2-e t h y l p h e n y l h y d r a z i n e h y d r O c h l O r i d e .C h i n e s e J O u r n a l [2] [3] [7] [8] [4][5] [9] [6] [10]妥妥妥妥妥妥妥、信息与交流节约能源!!!日本用绿地和风化解 "热岛# 现象受人类生产和生活的影响 人口密集的大都市会出现 G 热岛7 现象 东京平均气温在过去100年间平 均上升了3 摄氏度0G 热岛7 现象使炎热天气增多 空调使用量大幅增加 用电高峰数值不断突破 为了节省能源 日本正在研究利用绿地和自然风为 G 热岛7 降温0面积为58 公顷的新宿御园可以说是东京的 G 制冷空调7 首都大学东京的三上岳彦教授测量结果表 明 由草地和绿树掩映的新宿御园比周围平均低2 摄氏度 白天树木遮天蔽日 绿草和树叶蒸散水分起 到散热作用 风把冷气送到别处 使周围的温度下降0 无风的夜晚 冷气以每秒20 至30 厘米的速度向周 围荡漾 给周围80 至90米范围送去凉爽0 日本环境省正以新宿御园为范本进行模拟实验 给城市规划提供参考 研究大楼的位置 住宅的形状 怎样配置才能最大限度地利用绿地的冷气0日本大城市大多临海 为此 日本正在研究利用凉爽的海风为 G 热岛7 降温 缓解 G 热带夜7 <夜间 最低气温高于25 摄氏度> 现象0 三上教授去年在调查东京临海地区的气温分布时发现 受挡住海风入口 的汐溜高楼群的影响 处于下风口的新桥等地 和海滨相比温度平均高2 摄氏度0 楼群的南侧 海风沿古 川刮入 对这一带有抑制气温上升的作用0日本建筑研究所的足永靖信研究员利用超级计算机 对东京临海地区温度分布和风的流动进行模拟 发现因高楼 街道 公园和河流的影响 温度和风力会出现复杂的变化0为了验证 G 风路7 国土交通省计划今年夏天在东京站 汐溜 新桥等150 个地点 48 小时内连续观 测风向 风速和温度等 并与气象厅 环境省的数据对比 了解海风的规律0日本工业大学成田建一教授说 要充分利用海风 在建筑物的形状上下工夫很有必要0 在东京 名古 屋等临太平洋的城市 河的两岸夏天有凉爽的海风 冬天有从山上吹来的冷风 如果从海这一面来看 两 岸的建筑成 G 八7 字 就可以做到夏天海风送爽 冬天防止冷风0国土技术政策综合研究所键屋浩司研究员认为 科学预测风路和绿地的效果 可以为制定化解 G 热 岛7 现象的对策发挥作用 进而节省大量能源0妥妥、妥妥、。
专利名称:一种2-氯-4-甲磺酰基苯甲酸的制备方法专利类型:发明专利
发明人:闫海生,尹荃,赵瑞强,黄瑞,任树杰,喻为福申请号:CN201010034131.3
申请日:20100118
公开号:CN102126996A
公开日:
20110720
专利内容由知识产权出版社提供
摘要:本发明首次公开了应用吡啶作为2-氯-4-甲磺酰基苯甲酸液相氧化MC型催化体系的催化活化剂,提出了一种2-氯-4-甲磺酰基苯甲酸的制备方法:向以脂肪族羧酸为溶剂的2-氯-4-甲磺酰基甲苯反应体系中加入钴-锰-溴-吡啶四元复合催化体系,通入含有氧分子的气体,升温至100~255℃,维持体系压力在0.5~3Mpa。
反应结束后,分离出固体产物,即为2-氯-4-甲磺酰基苯甲酸。
应用本方法制备2-氯-4-甲磺酰基苯甲酸,可以大大降低主催化剂用量并提高反应速度,从而降低生产成本、满足工业生产的要求。
申请人:中国中化股份有限公司,沈阳化工研究院有限公司,沈阳化工研究院设计工程有限公司地址:100031 北京市西城区复兴门内大街28号
国籍:CN
代理机构:沈阳科苑专利商标代理有限公司
更多信息请下载全文后查看。
2004 年2 月 Journal of Chemical Engineering of Chinese Universities Feb. 2004 文章编号:1003-9015(2004)01-0094-052-(4-卤代苯甲酰基)苯胺的合成研究戴立言, 吴彩娟, 陈英奇, 吴兆立(浙江大学材料与化工学院,浙江杭州,310027)摘要:研究了重要的有机中间体2-(4-卤代苯甲酰基)苯胺的通用合成路线,是以邻苯二甲酸酐为原料,与卤代苯(分别为氟苯、氯苯和溴苯)进行付-克反应得到羧酸;羧酸经酰氯化、酰胺化,制得邻苯甲酰基苯甲酰胺衍生物,然后再经过霍夫曼降解合成了标题化合物。
对主要步骤反应条件进行了优化,得出比较合理的条件为:付-克反应,催化剂:AlCl3,配料比:1:2.2:6.4(苯酐:AlCl3:卤代苯);同时使用卤代苯既作反应试剂,又作溶剂,未反应的卤代苯回收套用,使收率基本定量;酰胺化反应氨气流量:10mL⋅min−1,使反应得以室温进行,无需制冷;四步反应总收率可达70%。
关键词:2-(4-卤代苯甲酰基)苯胺; 2-(4-氟苯甲酰基)苯胺; 2-(4-氯苯甲酰基)苯胺; 2-(4-溴苯甲酰基)苯胺;中间体; 合成中图分类号:O621.3; TQ246.31 文献标识码:A1 前言2-(4-卤代苯甲酰基)苯胺(1)(X=F、Cl、Br)是重要的有机原料和医药中间体,可用于合成植物生长调节剂和园艺杀菌剂异菸碱酰替苯胺[1]和还原酶HMG-CoA的阻聚剂NK-104[2],也用于合成2-氨基-3-卤代苯甲酰基苯乙酰胺衍生物,用于治疗眼科发炎症状[3],还可用于合成4-卤代苯-2-哌嗪喹啉衍生物,目前又发现该类物质具有抗溃疡作用[4]。
其主要合成路线如下:收稿日期:2001-12-20; 修订日期:2002-05-30。
作者简介:戴立言(1971-),男,辽宁开原人,浙江大学副教授、博士。
通讯联系人:戴立言,E-mail: dailiyan@路线(ⅰ):X=F、Br该路线有文献报道,是以邻苯二甲酸酐为原料,与卤代苯在无水三氯化铝催化下进行付-克反应制得羧酸(2),羧酸再经过酰氯化、酰胺化,得酰胺(3),然后经过霍夫曼降解得到标题化合物(1),收率为65%。
该合成路线,反应步骤较短,反应条件比较温和,操作简便,但X=Cl 时的路线目前未见文献报道; 路线(ⅱ): 同路线(ⅰ)方法得到羧酸(2)后, 与叠氮化钠一步反应得到胺(1),X=F、Br有该合成路线报道。
该路线步骤较少,但叠氮化钠操作危险,容易爆炸,反应要求严格,而且该路线的收率也很低,X=Br为 1.9%;路线(ⅲ):以邻氨基苯甲酸为原料,经过氨基保护、酰氯化和与卤代苯进行付-克反应得到标题化合物(1)。
该路线反应收率低,X=F收率为49%,X=Br时,仅最后一步付-克反应收率为28%,X=Cl时收率未见报道;路线(ⅳ)和(ⅴ)(X=Cl):均以靛红酸酐为原料,原料价格贵,且难以获得;路线(ⅳ)最后一步付-克反应,副反应多,收率为44%;路线(ⅴ)过程要用到格氏试剂,操作要求严格,条件苛刻,收率为41%;路线(ⅵ)和(ⅶ)(X=Br):分别以对溴碘苯和对二溴苯为原料,均要先制成格氏试剂,再反应而得(1),条件要求苛刻,反应收率也不高,分别为45%和48%。
本文采用路线(ⅰ)作为合成标题化合物(1)的通用路线,即以邻苯二甲酸酐为原料,与卤代苯进行付-克反应得到羧酸(2),再经酰氯化、酰胺化,得酰胺衍生物(3),然后经过霍夫曼降解合成胺(1),并经过改进使得付-克反应能进行完全,收率高达93%以上;酰胺化反应条件更温和,操作更为简便,在室温即可进行,四步反应收率可达70%,且该化合物X=Cl时,也顺利用此方法进行了合成。
2 实验部分2.1 化学试剂邻苯二甲酸酐(AR,江苏宜兴市第二化学试剂厂);氟代苯(CP,辽宁省义县精细化工总厂);氯代苯(CP,上海永华特种化学试剂厂);溴代苯(CP,上海青浦合成试剂厂);三氯化铝(AR,金山区兴塔美兴化工厂);甲苯(AP,杭州化学试剂有限公司);氯化亚砜(CP,上海亭新化工试剂厂);次氯酸钠水溶液(杭州电化集团公司)。
2.2 分析仪器Hectrothermal 9200IA型熔点仪,HP5989A质谱仪,日本岛津LC-10A液相色谱仪,ADVANCE DMX500型核磁共振仪,TMS为内标。
2.3 实验2.3.1 2-(4-氟苯甲酰基)苯甲酸的合成将25g(0.17mol)邻苯二甲酸酐,50g(0.375mol)无水三氯化铝加到100mL(1.08mol)氟苯中,反应立即进行,放出氯化氢气体,反应混合物此时呈桔黄色。
缓慢加热至75℃并保持,继续反应2h,冷却至室温,在搅拌下将红色反应液缓慢倒入冰水中,加入稀硫酸调节pH至1;减压蒸馏回收氟苯(76.2mL,0.82mol),回收完毕后冷却至常温,有大量白色粉末状固体析出。
抽滤,烘干得2-(4-氟苯甲酰基)苯甲酸38.5g,收率:93.8%,熔点:135~137℃。
(文献收率:90%,熔点:137~137.5℃)。
同方法可得2-(4-氯苯甲酰基)苯甲酸(收率:96.8%,熔点:142~144℃;文献收率:95%)和2-(4-溴苯甲酰基)苯甲酸(收率:93.3%,熔点:167~168℃;文献[16]收率:86%,熔点167℃)。
2.3.2 2-(4-氟苯甲酰基)苯甲酰胺的合成将20g(0.0823mol)2-(4-氟苯甲酰基)苯甲酸加到84mL甲苯中,再加入1mL DMF。
加热搅拌,缓慢滴加11.3g(0.0946mol)氯化亚砜,0.5h内滴加完毕,固体完全溶解并有气体放出,保温40~50℃反应4h,当不再有气体放出时减压蒸馏,回收溶剂及氯化亚砜至干。
再加入100mL甲苯,搅拌剧烈,室温下通入氨气,立即产生大量白色固体;继续通氨2h,停止反应,冷冻至0℃;减压抽滤,烘干得白色粉末状粗品,母液蒸馏回收甲苯。
粗品用100mL水搅拌20min,抽滤,烘干得2-(4-氟苯甲酰基)苯甲酰胺18.9g,收率:94.3%,熔点:170~172℃。
(文献收率:92%,熔点:177~178℃)。
同方法可得2-(4-氯苯甲酰基)苯甲酰胺(收率:94.4%,熔点:189~191℃)和2-(4-溴苯甲酰基)苯甲酰胺(收率:90.3%,熔点:214~215℃;文献[16]收率:90%,熔点213~215℃)。
2.3.3 2-(4-氟苯甲酰基)苯胺的合成将20g(0.0826mol)2-(4-氟苯甲酰基)苯甲酰胺加到90mL10%的氢氧化钠水溶液中,冰水浴冷却,使温度保持在0~5℃。
滴加53.8g(0.0866mol)12%的NaClO水溶液,在30min内滴完,固体逐渐溶解。
当溶液变成澄清时,将混合液缓慢滴入50mL75℃水中,立即有黄色固体产生;继续反应1h,冷却至室温,减压抽滤,滤饼用100mL水洗,烘干得黄色粗品, 用无水乙醇重结晶,并加活性炭脱色,得到浅黄绿色针状晶体2-(4-氟苯甲酰基)苯胺15.3g。
收率:86.6%,熔点:126~127℃,纯度(HPLC):99.77%。
(文献收率: 85.9%, 熔点: 127~128℃), MS(m/e): 238(M+Na,7.2%),216(M+H,100%);1H-NMR(CDCl3) δ(ppm): 5.98(2H,br,NH2),6.59~6.63(1H,t),6.73~6.75(1H,d),7.11~7.15(2H,t),7.26~7.31(1H,m),7.40~7.42(1H,d),7.66~7.68(2H,q)。
同方法可以制得淡黄绿色针状晶体2-(4-氯苯甲酰基)苯胺(收率:85.7%,熔点:99~101℃,纯度(HPLC):99.43%;文献熔点:98~99℃), MS(m/e): 254(M+Na,5.2%),232(M+H,100%);1H-NMR(CDCl3)δ(ppm):6.04(2H,br,NH2),6.58~6.62(1H,t),6.72~6.74(1H,d),7.25~7.30(1H,q),7.38~7.43(2H,q),7.57~7.58(2H,d), 黄绿色针状晶体2-(4-溴苯甲酰基)苯胺(收率:85.7%,熔点:109~110℃,纯度(HPLC):99.25%;文献[16]收率:84.0%,熔点106~107℃), MS(m/e): 300(M+Na,20.8%),278(M+H),100%);1H-NMR(CDCl3)δ(ppm):6.08(2H,br,NH2),6.58~6.61(1H,t),6.72~6.74(1H,d),7.26~731(1H,m),7.38~7.39(1H,d),7.50~7.52(2H,d),7.58~7.60(2H,d)。
3 结果和讨论3.1影响收率的因素付-克反应中,影响反应收率的因素有原料配比、反应温度和催化剂用量,今分别讨论如下,具体见表1,表2和表3。
3.1.1 原料配比对反应收率的影响按文献[5]投量(邻苯二甲酸酐:卤代苯=1:1.9),发现反应液会很快固化,难以再继续反应,根本达不到文献的收率,所以本实验中增加了卤代苯的投料量(配料比为1:6.4),使之即作为反应试剂又作溶剂,过量的卤代苯再回收利用,取得了很好的效果,收率可达90%以上,如果加上操作和后处理的损失,收率基本定量。
结果如表1所示。
表1 配料比对反应收率的影响T able 1 Effect of reaction mole ratio on the yieldReactions mole ratio 1:1.9 1:3.8 1:5.7 1:6.4 1:6.8 The appearance of reaction Quickly solidified Easily solidified Good Quite good Quite good The yield of (2) / % 32.6 72.5 87.5 93.8 94.2The reaction condition: 75℃; o-phthalic anhydride 0.17mol; anhydrous aluminum chloride 0.375mol.3.1.2 反应温度对反应收率的影响由表2可见,反应温度控制在75℃左右为宜,它对反应收率没有明显的影响,但对反应时间有着直接的关系,反应温度越高(<80℃)反应越剧烈,所需时间越短。
表2 反应温度对反应收率的影响Table 2 Effect of the reaction temperature on yield of (2)Number 1 2 3 4 Reaction temperature / ℃55 65 75 80 Reaction time / h 6.2 3.1 1.6 1.5The yield of (2) / % 90.7 91.4 93.8 93.1The reaction condition: o-phthalic anhydride 0.17mol; anhydrous aluminum chloride 0.375mol; fluorobenzene 6.4mol.3.1.3 催化剂用量对反应收率的影响参照文献[17]对三氯化铝用量进行了研究,由表3可见,对反应收率影响最大的因素是催化剂三氯化铝的用量,当摩尔投料比为1:2.2(邻苯二甲酸酐:三氯化铝)得到较理想的收率和较短的反应时间。