ISO9001-ISO14001-TS16949一体化审核指南
- 格式:ppt
- 大小:1.78 MB
- 文档页数:8
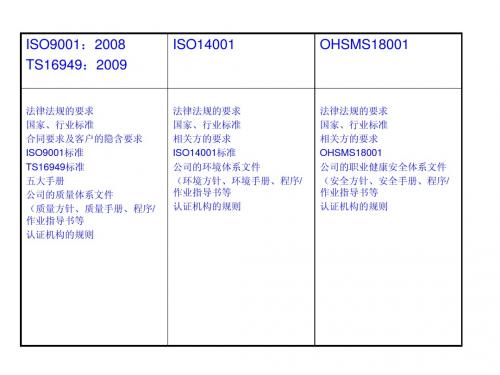

提到管理体系,大家都会不由自主地想到ISO9001、ISO14001、OHSAS18001、ISO/TS16949、 TL9000等,其实这些体系都是有区别的。
下面,就其中的两种--ISO9001和ISO14001,给大家分享一下具体的区别,以便大家进行参考。
一、定义不一样
ISO9001是治理管理体系,旨在质量这一方面,对生产行业,从原料到生产过程到FQC的一个严格的把控;对于服务行业,就是服务水平和服务质量的管理。
ISO14001是环境管理体系,顾名思义,就是对生产或者服务过程中可能涉及环境问题进行一个管理和控制。
二、内部审核时间的规定不一样
ISO9001质量管理体系标准要求按策划时间间隔;ISO14001环境管理体系标准要求按计划的时间间隔。
三、对其的要求不一样
质量体系要满足质量管理和对顾客满意的要求,环境管理体系要服从众多相关方的需求,特别是法规的要求。
以上三点就是这两者之间的一些区别,希望能帮到大家。

ISO9001审核实践指南国际认可论坛日期: 2005年12月12日ISO9001审核实践小组是由从质量管理和质量保证技术委员会(ISO/TC176)和国际认可论坛(IAF)遴选出来的质量管理体系(QMS)专家、审核员和从业者组成的非正式工作组。
该工作组编写了一系列包含了有关QMS审核的理念、实例和解释的指南文件(见下附“QMS审核主题”)。
这些文件体现了基于过程的方法,过程方法是审核ISO9001:2000质量管理体系——要求的精髓。
本指南主要供但不限于QMS审核员、咨询师和质量从业者使用。
这些文件反映了QMS审核的一些不同观点。
因此,这些文件的内容并不总是一致的。
本指南不期望被用作规定的要求、行业基准、或作为所有QMS审核员、咨询师或从业者必须遵守的准则。
QMS审核主题·两阶段审核方式的必要性·测量QMS有效性和改进·过程的识别·理解过程方法·“适当时”过程的确定·审核“适当时”要求·证实与标准的符合性·特定任务、活动或过程的审核与整个体系相联系·审核持续改进·审核具有最低限度文件的QMS·如何审核最高管理者过程·审核检查表的作用和价值· ISO9001:2000的范围、质量管理体系(QMS)的范围和确定认证的范围·如何在审核过程中增值·审核“能力”和“所采取措施的有效性”·审核法律法规要求·审核质量方针和质量目标·审核ISO9001,条款监视和测量装置控制·有效使用ISO19011·审核顾客信息反馈过程·出具不符合项·评审及关闭不符合项的指南·审核内部沟通·审核预防措施·审核服务组织·第三方审核员公正性和利益冲突·审核内部审核有效性·审核电子化管理体系ISO9001审核实践小组将根据使用者的意见反馈决定是否编制补充的指南文件或对现在的文件进行修订。

XX公司环境管理体系内部审核计划
一、审核目的
评价公司的环境管理体系是否符合ISO14001:2015标准以及公司的要求,是否得到有效的实施和保持。
二、审核范围
1.XX产品的设计开发、生产、销售及相关的管理活动
2.地点:广东省佛山市顺德区XX路XX 号
3.审核取证时间:2017年3月20日至审核时止
三、审核类型
例行内部审核。
四、审核准则
1.ISO14001:2015标准
2.公司的环境地管理体系文件
3.适用的法律法规及其他要求、合同
五、现场审核时间
2018年3月17~18日
六、审核组
审核组长:陈XX(A)
审核组员:李XX(B)、张XX(C)、王XX(D)
七、审核要求
1.审核期间公司各部门负责人应参加首次会议和末次会议。
2.审核期间被审核部门的有关人员应根据审核的安排,在相应的工作岗位上。
3.审核计划规定的是大致时间,可能根据审核需要作相应调整。
八、审核日程安排
编制:陈XX 2018年3月7日审核:李XX 2018年3月7日批准:张XX 2018年3月10日。


年审ISO9001:2008所必备的资料:1、ISO9001体系文件一套;认证合同2、质量目标的统计、分析及没有完成目标部门的整改报告(原因分析、纠正和预防措施、验证);3、ISO9001内审资料(内审计划、检查表、签到表、不符合项报告、内审报告、不符合项报告的结案);4、ISO9001管理评审资料(管理评审通知,管理评审报告);5、客户满意度调查、分析及改进;6、合同评审相关记录;7、年度培训计划及相关签到记录;8、供应商的评审计划及评审记录;9、上次认证审核的所有资料;10、用于产品检验和试验的仪器、工具的内外校报告、清单;11、公司的营业执照,机构代码复印件;12、各部门运行的相关记录提供。
年审ISO13485:2003所必备的资料:1、ISO13485体系文件一套;程序文件中必须要有:风险管理控制程序,反馈系统控制程序,忠告性通知、发布和实施程序;应急的准备和响应控制程序;计算机软件确认程序和灭菌过程确认程序(不适用富力达);不良事件告之行政主管部门的程序(法规有要求时必须要);2、ISO9001:2008证书复印件;ISO13485认证合同3、营业执照和机构代码复印件;税务登记证;4、每一款医疗器械产品的风险评估报告;5、ISO13485内审资料;6、ISO13485管理评审资料;7、上次认证审核ISO13485的所有相关资料;8、ISO13485目标的统计、分析及没有完成目标部门的整改报告(原因分析、纠正和预防措施、验证);9、做医疗器械相关部门的作业环境规定,制造过程和返工的SIP/SOP;相关记录(保存期限10年) 提供。
10、医疗器械产品法律法规的搜集及符合性评价。
年审QC080000:2005所必备的资料:1、QC080000体系文件一套;认证合同2、ISO9001:2008证书复印件;3、营业执照和机构代码复印件;4、QC080000目标的统计、分析及没有完成目标部门的整改报告(原因分析、纠正和预防措施、验证);5、QC080000内审资料;6、QC080000管理评审资料;7、上次QC080000认证审核的所有相关资料;8、ROHS检测清单;检测计划9、HS清单及标准;相关产品的环保法律法规清单及符合性评价。