03-BLAST(生物信息学国外教程2010版)
- 格式:ppt
- 大小:6.23 MB
- 文档页数:123

南方医科大学实验报告姓名学号专业年级基础学院生物信息学教研室题目BLAST 日期实验者实验者一、实验目的一、实验目的1,了解BLAST算法原理算法原理2,掌握BLAST参数设定的意义参数设定的意义3,利用BLAST解决生物学问题,如寻找给定序列(DNA或者蛋白质)的同源序列。
或者蛋白质)的同源序列。
二、实验器材二、实验器材电脑电脑三、方法与步骤三、方法与步骤)或者用自己的序列。
给定人蛋白RBP4(NP_006735)或者用自己的序列。
1 限定物种为人(Homo sapiens ),在参考序列数据库中搜索,列出结果(具体比对不列)。
1)进入BLAST主页主页/Blast.cgi2)限定物种为人Homo sapiens 3)在参考序列数据库中搜索在参考序列数据库中搜索4)CLICK BLAST 2 分别限定物种为Mus musculus ,Rattus norvegicus ,Drosophila melanogaster ,Bos taurus ,Danio rerio,各列出E值最小的两条序列。
值最小的两条序列。
1)选择物种选择物种3)输入序列,选择数据库和BALST程序,点击BLAST 4)E值最小的两个序列值最小的两个序列5)其余物种依次重复其余物种依次重复四、结果与讨论四、结果与讨论限定物种为人(Homo sapiens ),在参考序列数据库中搜索,列出结果(具体比对不列)。
分别限定物种为Mus musculus ,Rattus norvegicus ,Drosophila melanogaster ,Bos taurus ,Danio rerio,各列出E值最小的两条序列。
值最小的两条序列。
Mus musculus Rattus norvegicus Drosophila melanogaster Bos taurus Danio rerio 。


BLAST使用方法一、BLAST的安装和准备工作2.获取待比对的序列文件,可以是FASTA格式的DNA或蛋白质序列。
二、BLAST的常用参数和选项1. Program:指定使用哪种BLAST程序(如BLASTn、BLASTp等)。
2. Database:指定使用哪个数据库进行比对。
3. Query:指定待比对的序列文件。
4. E-value:期望值。
一种描述比对结果误差率的指标,值越小表示结果越可信。
通常情况下,E-value小于0.01被认为是显著结果。
5. Word size:BLAST在比对时使用的核心词的长度。
长度越大表示查全率(sensitivity)越高,但速度会减慢。
6. Gap open:允许在比对过程中插入空位(如插入一个碱基)。
Gap open参数定义了开放一个空位的惩罚分数。
7. Gap extension:允许空位的延伸。
Gap extension参数定义了延伸一个空位的惩罚分数。
三、使用BLAST进行比对1.命令行方式:-打开命令行界面,并定位到BLAST软件的安装目录。
- 输入命令,指定BLAST程序、数据库、查询文件和其他参数。
例如:blastn -db nt -query query.fasta -out output.txt -evalue 0.01-运行命令,BLAST将开始进行比对并生成结果文件。
2.网页方式(以NCBIBLAST为例):- 打开NCBI网站的BLAST页面()。
-选择需要使用的BLAST程序(如BLASTn、BLASTp等)。
-上传待比对的序列文件,或者粘贴序列文本到输入框中。
-选择适当的数据库和其他参数。
-点击“BLAST”按钮,等待比对完成。
四、解读BLAST结果1. E-value:表示在随机比对中获得与查询序列相似度更高的结果的期望概率。
E-value越小表示比对结果越显著。
2. Bitscore:用于表示比对结果的质量。
Bitscore越高表示比对结果越可信。
实验三数据库搜索—BLAST1. Nucleotide BLAST在Nucleotide中输入登录号搜索人类MAPK9(NM_139069.2)基因,send to 为coding sequences,作为Query 序列,或者下载complete sequences,在Blastn 中限制序列搜索范围为272-1420(编码区)。
分别用megablast, discontiguous megablast 和 blastn 进行搜索。
这三个搜索的参数不同之处,主要体现在单词单位,megablast的单词单位默认为28,可选范围从16-256, discontiguous megablast的单词单位默认为11,可选为11和12, blastn单词单位默认为11,可选范围为7,11和15。
Megablast 可以快速搜索到与query 高度相似的序列;discontiguous megablast用于寻找与 query 高度相似的序列; blastn则用于寻找与 query 有一定相似度的序列。
单词单位越小,敏感度越高,也就是说,Megablast敏感度最差,discontiguous megablast 居中,blastn 最高。
Megablast的搜索速度最快,discontiguous megablast居中,blastn最差。
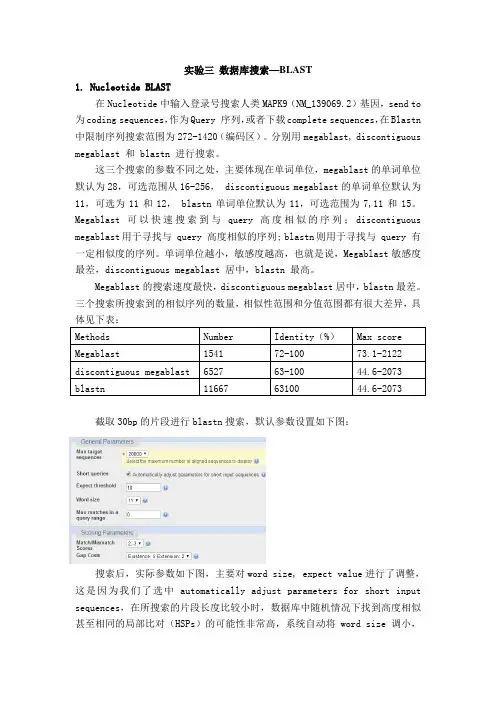
三个搜索所搜索到的相似序列的数量,相似性范围和分值范围都有很大差异,具Methods Number Identity(%)Max score Megablast154172-10073.1-2122 discontiguous megablast652763-10044.6-2073 blastn116676310044.6-2073截取30bp的片段进行blastn搜索,默认参数设置如下图:搜索后,实际参数如下图,主要对word size, expect value进行了调整,这是因为我们了选中automatically adjust parameters for short input sequences,在所搜索的片段长度比较小时,数据库中随机情况下找到高度相似甚至相同的局部比对(HSPs)的可能性非常高,系统自动将 word size 调小,提高敏感度,而将 E-value 调大,确保有搜索结果出现。





BLAST种类及使用方法BLAST(Basic Local Alignment Search Tool)是一种广泛使用的序列比对算法,可用于比较DNA,RNA或蛋白质序列的相似性。
它是生物信息学领域中最常用的工具之一,可以帮助研究人员识别新的序列,注释基因功能,鉴定物种间的进化关系等。
1.BLASTN:BLASTN用于比对DNA序列。
它可以将一个查询DNA序列与已知的DNA序列数据库进行比较,找到相似的序列。
BLASTN通常用于物种鉴定、基因组注释和寻找同源基因等方面的研究。
2.BLASTP:BLASTP用于比对蛋白质序列。
它可以将一个查询蛋白质序列与已知的蛋白质数据库进行比较,找到相似的蛋白质序列。
BLASTP 通常用于寻找同源蛋白质,预测蛋白质功能和结构,以及识别蛋白质家族等方面的研究。
3.BLASTX:BLASTX用于比对DNA序列与蛋白质数据库的比对。
它通过将DNA序列翻译成蛋白质序列,然后与已知的蛋白质数据库进行比对,找到相似的蛋白质序列。
BLASTX通常用于从未知的DNA序列中预测蛋白质编码区域,注释基因功能等方面的研究。
4. TBlastN:TBlastN用于比对蛋白质序列与DNA数据库的比对。
与BLASTX相反,TBlastN将已知的蛋白质序列与DNA数据库进行比对,找到相似的DNA序列。
TBlastN通常用于寻找蛋白质在基因组中的编码区域,确定启动子和转录因子结合位点等方面的研究。
5. TBlastX:TBlastX用于比对转录本与转录本数据库的比对。
它可以将一个查询转录本序列与已知的转录本数据库进行比对,找到相似的转录本。
TBlastX通常用于寻找新的转录本和预测基因表达模式等方面的研究。
使用BLAST有以下几个步骤:1.准备查询序列:将待比对的DNA、RNA或蛋白质序列准备成文本文件,确保序列格式正确,并确保序列长度适合比对任务。
2. 选择数据库:根据研究需求,选择适当的数据库。


2003年3月内蒙古大学学报(自然科学版)M ar.2003第34卷第2期Acta Scientiarum Naturalium Univ ersitatis NeiM ongol Vol.34No.2 文章编号:1000-1638(2003)02-0179-09生物信息学工具BL AS T的使用简介吕 军1,3,张 颖3,冯立芹2,李 宏1(1.内蒙古大学理论物理与理论生物物理研究室,内蒙古呼和浩特010021;2.内蒙古民族大学物理系,内蒙古通辽028043;3.内蒙古工业大学物理教研室,内蒙古呼和浩特010062)摘要:从网上在线服务、电子邮件服务和本地运行三个方面介绍BL AS T的使用方法,目的是使大家尽快掌握它,使其成为理论生物学研究的有力工具.关键词:BL AS T;数据库;搜索中图分类号:Q617 文献标识码:A引 言 随着人类基因组计划(HGP)的进展,生物数据量迅速膨胀,海量的生物数据摆在生物信息学的工作者面前.生物信息学计算的核心是序列的比较,从而,比较基因组学、比较蛋白质组学成为后基因组时代的主要研究方向之一.比较的内容从序列的组分变化、寻找特殊的字段,到序列间字母的对应.比较的主要目的在于阐明序列间的同源(isogeny)关系,以及从已知序列去预测新序列的结构和功能.两个或多个符号序列按字母比较,尽可能确切地反映他们之间的相似和相异,称为序列的联配(a lig nment).核酸和蛋白质序列的联配的前提是,假定两个序列来自同一个祖先序列(“同源”),它们在演化的过程中由于变异的积累而成为不同的序列.近年来,进行序列联配分析的工具软件发展了很多,其中,尤以BLAST和FAST A使用最为频繁,一般认为,BLAS T运行速度快,对蛋白质序列的搜寻更为有效,FASTA速度较慢,对核酸序列更为敏感.BLAST是“基本局域联配搜索工具”(Basic Local Alig nment Search Tool)的字头缩写,是最常用的比较核酸和蛋白质同源性的比较工具.现在,利用BLAST对数据库进行搜索已成为生物信息学工作者的经常.因为BLAST和FAS TA的功能相近,所以,本文以BLAS T为例从三个方面来分别介绍BLAST的使用方法.关于BLAST的算法描述可见文献〔1〕和〔2〕.1 网上在线服务 BLAST是运行速度甚快的数据库搜索程序,许多生物信息中心都有专门运行BLAST的服务器.主要的BLAST服务器网址如下:http://w w w.ncbi.nlm.nih.g ov/blast/(运行BLASTR2.0,美国,维护GenBank)http://w w (运行W U-BLAST2,欧洲,维护EM BL数据库)http://w w w.blast.geno me.ad.jp/(运行BLAST2.0,日本)(运行BLASTR2.0,中国,有ncbi和ebi的镜像)收稿日期:2002-05-17基金项目:国家自然科学基金(10147204)资助项目,内蒙古自然科学基金(2001301)资助项目作者简介:吕军(1973~),男,内蒙古乌拉特前旗人,讲师,硕士.各服务器的BLAS T 搜索界面大同小异,下面我们以CBI(北京大学生物信息中心)的BLAS T 服务器为例,分步骤来介绍BLAST 的在线搜索方法.第一步:首先以http :// 登录CBI 的BLAS T 服务器,其界面如图1,这时可以选择所要进行的搜索方式.主要的搜索方式列在表1中.其中,BLAST 2Sequences 只针对两条序列的比对.从表1中可以看出,在BLAST 前面加T 表示要求进行翻译,后面加N 、P 分别表示核酸和蛋白质库.X 则表示某种交叉比较.图1 CBI 的BL A ST 程序选择界面Fig.1 H o me Pag e of Pro g ram o f BL A ST o f CBI表1 BLAST 程序Table 1 Programs of BLAST Prog ra ms Query Sequences Sea rch Data Bases BL A ST N Nucleotide N ucleotide BL AS T P Pro tein Pro tein BL A STX Nucleotide Pro tein T BL A ST N Pro tein N ucleotide T BL AST X NucleotideN ucleotideBL A ST 2Sequences 第二步:根据需要选择一种搜索方式后,进入下一层界面,如图2(以BLASTN 为例).这就是BLAS T 的主界面,下面我们逐一介绍一下BLAS T 的主界面中的各选项.图2 BL A STN 查询主界面Fig .2 Quer y Hom e Pag e o f BL A ST NA.首先是选择数据库 核酸序列数据库和蛋白质序列主要数据库分别列在表2中.对数据库的选择可根据查询的具体要求做出相应的选择.缺省数据库为nr.180内蒙古大学学报(自然科学版)2003年表2 蛋白和核酸数据库Table 2 Pept ide and Nucleotide Sequence DatabasesPeptide Sequence Databasesnr All no n-redundant GenBa nk CD S t ransla tions +PDB +SwissPr ot +PI RmonthAll new or revised GenBa nk CD S tra nsla tio n +PDB +Sw iss Pro t +PIR released in the last 30days .Swisspro t The last major release of the SW I SS-PRO T pro tein sequence database (no upda tes)y ea st Yeast (Saccharo myces cerevisiae )pro tein sequences.E .co li E .co li geno mic CDS tra nslatio nsPdbSequences deriv ed fr om the 3-dimensional structur e Br oo khav en Pro tein Da ta Ba nkPat Pro tein sequences deriv ed fro m the Pa tent divisio n o f G enBank N ucleo tide Sequence Da ta ba sesnr All N on -redunda nt GenBa nk +EM BL +DDB J +PDB sequences (but no EST ,ST S ,G SS ,or HT GS sequences)est N o n-redundant Database of GenBank +EM BL+DDBJ EST Div isionssts N o n -redundant Database of GenBank +EM BL +DDBJ ST S Div isions h tg s H ig h Thro ughput Genomic SequencesgssGeno me Surv ey Sequence ,includes sing le -pass g eno mic data ,ex o n -tr apped sequences ,a nd Alu PCR sequences.B .输入FAST A 格式的要查询序列 需要查询的序列可以通过“查询序列输入文本框”提交,也可以将要查询的序列做成FAST A 格式的文件通过“查询序列文件载入文本框”提交.这里允许同时提交多个序列.要求需要查询的序列必须以FAS TA 格式录入.FASTA 格式以“>”开头,每行不超过80个字符(包括说明行).最好每行低于60个字符.FAST A 格式支持的核酸符号和氨基酸符号分别列在表3中.表3 FASTA 格式所支持的核酸符号和氨基酸符号Table 3 The nucleic acid codes and amino acid codes supported by FASTAThe nucleic acid codes suppo r ted a re A adeno sine M A C (a mino )U uridine D G A T C cytidine S G C (stro ng )R G A (purine )H A C T G guanineWA T (weak)Y T C (pyrimidine)V G C A T thymidineB G T CKG T (keto )NA G C T (a ny )*ga p o f indetermina te leng thThe amino acid co des suppo rted ar e A a la nine G g lycine N a spar agine U selenocy steine B a spar tate o r asparag ine H histidine P pro line V v aline C cystine I isoleucine Q glutamine Wtr yptophanD a spar tate K lysine R a rginine Y tyr osineE g lutama te L leucineS serine Z g lutama te or glutamine F phe ny lalanine M me thionineTth reo nineXany*tr anslatio n stop-ga p o f inde termina teC.过滤程序的选择缺省为低复杂度,过滤程序可以滤掉序列中的一些“低复杂度”区域,否则象Poly (A)、Poly (T)这样的片段会导致高分联配,漏掉真正的编码区.一般选取缺省值即可.BLASTN 只能选用或不用DU ST 过滤程序,其他可选用过滤程序为SEG 、XN U 或其组合.对于这些过滤程序的描述可参见〔3〕等文献.D.期望值E 的选择181第2期吕军等 生物信息学工具BLAS T 的使用简介期望值E是一个统计显著性指标,是假定所提交的序列和库中的全部序列都是随机序列,所期望的符合数目.只有搜索到比期望值小的符合序列,才作为结果返回.缺省为10.E.联配矩阵的选择连配打分矩阵的选择的一般原则见表 4.F.交叉搜索时遗传密码表的选择和移框的选择(仅对BLASTX)G.高级选项高级选项的参数及其缺省值得设置见表5.高级选项的使用可以增加查询的灵活性,建议大家在对BLAST比较熟悉时,一定去使用这些选项.表4 打分矩阵的选择原则Table4 C hoice principle of substitut ion matrixQ ue ry leng th Substitutio n matrix <35P AM-3035-50P AM-7050-85BL O SU M-80>85BL O SU M-62表5 BLASTN和BLASTP、BLASTX、TBLASTN高级选项Table5 Advanced Options of BLASTN,BLASTP,BLASTX and TBLASTN BL A ST N Adv anced O ptio ns-G Cost to o pen a ga p[Intege r]default=5-E Cost to ex tend a g ap[Integ er]default=2-q Penalty for a mismatch in the bla st po rtion of run default=-3-r Rew ar d fo r a ma tch in th e blast por tio n o f r un defa ult=1-e Ex pecta tio n va lue(E)[Real]defa ult=10.0-W W ord size,default is11for blastn,3fo r o th er pro g rams.-v N umbe r o f o ne-line descriptio ns(V)[Integ er]defa ult=100-b N umbe r o f a lig nments to show(B)[Integ er]default=100BL A ST P,BL AST X,TBL A ST N Adv anced O ptio ns-G Cost to o pen a ga p[Intege r]default=11-E Cost to ex tend a g ap[Integ er]default=1-e Ex pecta tio n va lue(E)[Real]defa ult=10.0-W W ord size,default is11for blastn,3fo r o th er pro g rams.-v N umbe r o f o ne-line descriptio ns(V)[Integ er]defa ult=100-b N umbe r o f a lig nments to show(B)[Integ er]default=100H.返回结果的浏览形式I.返回符合序列简短说明的行数.缺省为100和50J.返回联配结果的颜色方案.共7种方案,缺省为无颜色方案K.清除查询序列和确定搜索第三步:分析返回结果.单击搜索按钮后,稍作等待便可有返回结果.返回结果主要包括以下四个部分,表10给出一个详细例子.A.标题(Header)A.1本次查询所使用的程序和其版本.A.2本程序的作者以及参考文献.A.3本次查询所搜索的数据库,搜索的序列数及字符数.A.4所提交的查询序列的信息.B.摘要(Summa ry)满足查询条件的有意义的联配结果的摘要信息.C.主要部分(Main)满足查询条件的有意义的联配结果详细信息.D.最后部分(finality)182内蒙古大学学报(自然科学版)2003年列出本次查询所使用的参数,以及一些计算出的参数.2 电子邮件查询服务 在线查询不方便时,比如受到上网机时的限制或所要提交的序列较长,可以通过电子邮件的方式进行BLAS T 搜索服务.因为是用电子邮件提交查询序列,故而没有上网费用的限制,所以最好提交国外网站(比如NCBI ),因为那里的数据库是最新最全的.下面我们就以NCBI 的电子邮件BLAS T 服务为例来演示如何通过电子邮件提交查询序列.我们发往N CBI 这样一封信: From :lujun8210@ Date :5Apr 200200:10:02(可选) To :blast @ Subject : PRO GRAM blastn DATALIB nr EX PEC T 0.75(可选) BEGIN >gi |6226515|ref |NC 001224.1|Saccharomy ces cerevisiae mitocho ndrion , complete g eno meT TCAT AAT TAAT T TT TT ATAT AT ATAT TA TAT TAT AAT ATT AATT T A ……其余参数均使用缺省值.返回结果与CBI 在线服务的返回结果基本相同,具体可参见表10.这里不再重复.3 本地运行 除在线BLAS T 搜索和E -M AIL 提交外,还可以将BLAST 下载到本地计算机上运行.下面我们分步骤来介绍如何在本地计算机上使用BLAS T 进行序列的比对.这对于我们的工作是十分有帮助的.第一步:下载一个单机用的DOS 版的BLAST 程序.可以到NCBI 下载(ftp ://ncbi .nlm .nih .g ov /blast /),也可以到谈杰的生物软件网下载(h ttp ://w w w.bio-so ).文件名为blastz.ex e,这是一个2.2版的BLAS T 程序,程序大小为7.3Mb.第二步:安装BLAST将blastz .ex e 拷贝到一指定目录(比如C :\Blast ,后面的演示均以此目录为准)后,运行blastz .ex e ,此时,在这一目录中生成12个应用程序、6个说明文件和一个data 文件夹.第三步:创建ncbi.ini 文件.在你的操作系统安装目录(如c :\w indow s)下创建一个名为ncbi.ini 的配置文件,在这个文件中写入下面两行代码,如果此文件存在,则修改其内容为下面两行代码: [NCBI] Data="path \data \" 比如,本例中在c :\window s \ncbi .ini 文件中写入下面两行代码: [NCBI ] Data=c :\blast\data因为在data 目录下存放有搜索必需的矩阵、密码表、参数表以及一些C 语言的脚本程序等重要信息,所以在搜索前要指定这些文件的存放路径.这一步做完之后,接下来要完成重要的一步,就是格式化数据库.第四步:格式化数据库.183第2期吕军等 生物信息学工具BLAS T 的使用简介184内蒙古大学学报(自然科学版)2003年首先要创建FASTA格式的数据库文件,然后用fo rm atdb程序将所创建的数据库文件格式化.下面我们详细介绍数据库的创建和格式化过程.A.创建数据库将所操作数据库以FAST A格式保存,可以自己创建,也可以到N CBI去下载,地址为:ftp:// /blast/db/.具体形式为:>gi|6319248|ref|N P009331.1|Yal069w pM IVNN T H V L TLPLYT TT TCHT HPHLY TDFTY AHGCYSIY HLKLTLL……....................................这里“>"是必需的,“>"之后是一些说明信息(说明可以省略),比如gi是NCBI数据库中序列的统一编号形式,gi后面跟“|”,“|”后面就是这个基因或蛋白的在NCBI的标识代码.具体标识代码含义见表6.包括说明在内,每行不超过80个字符,这是一个默认值.每个基因或蛋白均以“>”开头.将这个数据库文件以一定的文件名保存.比如创建一个核酸数据库以nt这个文件保存在“c:\blast\ database\nt\"下.表6 数据库名称与标识码Table6 Database Name and Identif ier SyntaxData base N ame Identifier Sy ntaxG enBank g b|accession|locusEM BL Data Libra ry emb|accessio n|locusD DBJ,DN A Database o f J apan dbj|accessio n|locusN BRF PIR pir||entr yPro tein Research Foundation pr f||na meSW ISS-PRO T sp|accessio n|entry na meBro okhav en Pro tein Data Bank pdb|entr y|chainPa tents pa t|countr y|numberG enInfo Backbone Id bbs|numberG ener al database identifier g nl|da taba se|identifie rN CBI Reference Sequence ref|a ccessio n|lo cusB.将所建好的数据库格式化启动DO S窗口,将路径指向BLAST应用程序所在目录,使用fo rmatdb命令对nt这个数据库进行格式化.fo rmatdb命令的语法格式为:fo rm atdb-i dtatbase-p F-o T本例中,可以如此应用fo rmatdb命令:c:\blast>fo rmatdb-i dtatbase\nt\nt-p F-o T其中一些主要参数的含义见表7.表7 f ormatdb命令的参数Table7 Parameters of f ormatdb commandPara meters sig nifica tio n-t Title for da ta ba se file[String]Optio na l-i Input file for for matting(this pa ramete r must be set)-l Log file name:O ptio nal defa ult=fo rma tdb.log-p T ype of file T-pr ot ein F-nucleo tide default=T-o Pa rse o ptionsT-Tr ue:Pa rse SeqId and cr ea te index es.F-False:Do not par se SeqId.Do no t cr ea te index es.[T/F]Optio na l default=F 执行完fo rm atdb命令后,在“c:\blast\database\nt\”下生成一系列文件,这些文件是进行数据库查询所必需的.第五步:进行序列比对和序列查询.完成以上步骤后就可以使用BLAST 进行序列的比对和查询了.A.两个序列之间的比对——bl2seq有时我们只需要对两个序列进行比对,此时就可以使用bl2seq 命令,使用bl2seq 命令不须要创建数据库,直接给定两条序列就可实现比对.具体语法格式为:bl2seq-i seq1-j seq2-p blast Pro gram-o out.Filebl2seq 命令还有很多参数,但最一般的是上面所列参数,根据所讨论问题的要求可具体选择不同的参数.查询参数的办法为,在提示符下直接键入bl 2seq ,不带任何参数即可,表8列出bl 2seq 命令的一些主要参数,注意参数的大小写.表8 bl 2seq 命令的参数Table 8 Parameters of bl 2seq command Pa ramete rssig nificatio n-i Fir st sequence [File In ]-j Second sequence [File In ]-p Prog r am na me :bla stp,blastn,blastx.For blastx 1st a rg ume nt sho uld be nucleo tide [String ];default =bla stp-o alig nm ent o utput file [File O ut ];default =stdout-M M a trix [String ];defa ult =BL O SUM 62-q Penalty for a nucleotide misma tch (blastn o nly );default =-3-r Rew ar d fo r a nucleo tide match (blastn o nly );default =1-e Ex pecta tio n va lue (E)[Real ];default =10.0-FFilter query sequence (DU ST w ith blastn,SEG w ith o ther s)[St ring ]default =T 举一个具体的例子,我们在“c :\blast da tabase \”目录下创建两个FASTA 格式的序列文件seq 1和seq 2,为了简单起见,我们把这两个文件内容作的相同.seq1:>gi |4001550|dbj |AB001390.1|AB001390Hepatitis C virus g ene for E2pro tein,hyperva riable re-gio n,pa rtial cds,clo ne :ACACACCC TCG TGAC AGGGGGGGseq2:>gi |4001550|dbj |AB 001390.1|AB 001390Hepatitis C virus g ene for E 2pro tein ,hyperva riable re-gio n ,pa rtial cds ,clo ne :ACACACCC TCG TGAC AGGGGGGG文件创建好之后,在DO S 窗口下输入以下命令行,输出文件名我们定义为seq12.c :\blast>bl2seq-i database\seq1-j da tabase\seq2-p bla stn -o da tabase\seq12-e 0.01命令执行后,我们察看输出文件seq12,如果-o 参数缺省,则查询结果在屏幕输出.seq12:Query :1cacaccctcg tg aca 15 |||||||||||||||Sbjct :1cacaccctcg tgaca 15 ............我们发现上面只比对了序列中的1-15个碱基,而16-22这7个碱基被滤掉了,因为这是连续7个G 的简单重复序列,如果不想滤掉它们,只须在bl2seq 的语句中加入参数-F,并且赋值F(假)即可,因缺省时-F 的值为T(真).即:185第2期吕军等 生物信息学工具BLAS T 的使用简介c:\blast>bl2seq-i database\seq1-j da tabase\seq2-p bla stn-o da tabase\seq12-e0.01-F F seq12:.................. Query:1cacaccctcg tgacaggg gg gg22|||||||||||||||||||||| Sbjct:1cacaccctcg tgacaggg gg gg22..................B.序列查询——blastall进行序列查询前,必须要有创建并格式化数据库的过程.前面的工作做好后,就可以利用blastall 命令进行序列的查询.具体语法格式为:blastall-p blastProg ram-d database-i Query File-o out.Query Fileblastall命令的部分参数列在表9中.寻求帮助时,直接键入blastall,不带参数就可以获得blastall命令的所有参数说明.注意参数的大小写区别.表9 blastall命令的参数Table9 Parameters of blastall commandPara meters sig nifica tio n-p Prog r am N ame[String]Input sho uld be one of"bla stp","blastn","blastx","tbla stn",o r"tblastx".-d Da ta base[String]default=nr-i Q uery File[File In]default=stdin-e Ex pecta tio n va lue(E)[Real]defa ult=10.0-o BL A ST repor t O utput File[File Out]Optio na l default=stdo ut-F Filter query sequence(DU ST w ith blastn,SEG w ith o ther s)[St ring]default=T-S Q uery stra nds to sear ch ag ainst da taba se(fo r blast[nx],a nd tblastx).3is bo th,1is to p,2is bo tto m [Integ er]default=3-T Produce HT M L output[T/F]default=F-l Rest rict sear ch o f da ta ba se to list of GI's[St ring]O ptio nal-U Use low er case filtering o f FA ST A sequence[T/F]O ptio nal defa ult=F 举一个例子.比如我们要在前面格式化好的数据库"c:\blast\da tabast\nt\nt"中搜索上例中的seq1这个序列.我们可以这样做:c:\blast>blastall-p blastn-d database\nt\nt-i database\seq1-o database\out.seq1-e0. 01-F F 输出文件为o ut.seq1,如果-o参数缺省,则查询结果在屏幕输出,具体结果见表10.表10 BLAST结果Table10 BLAST resultBL A ST N2.2.2[Dec-14-2001]Header Ref erence:Altschul,Stephe n F.,Thoma s L.M adden,Alejandro A.Schaffer,Jingh ui Zhang,Zheng Zhang,W ebb Miller,and Dav id J.Lipman(1997),"Gapped BL A ST and PSI-BL A ST:a new g eneration of pr otein database searchpro g rams",N ucleic Acids Res.25:3389-3402.Query=gi|4001550|dbj|AB001390.1|A B001390H epatitis C v irus gene fo rE2pr o tein,hyperv ariable regio n,pa rtia l cds,clo ne:A(22letter s)Da taba se:da ta ba se\nt\nt1386sequences;2,070,001to tal let ters186内蒙古大学学报(自然科学版)2003年续表10Sco r eE Sequences pro ducing sig nifica nt a lig nments :(bit s)V alue Summa rydbj |AB001409.1|AB001409Hepatitis C v irus g ene fo r E2pr otein,h (4)1e-006........................>dbj |AB001409.1|AB001409Hepa titis C virus g ene fo r E2pro tein,hy perv ariable regio n, pa rtia l cds ,clo ne 18A L eng th =81Sco r e =44.1bits (22),Ex pect =1e -006Identities =22/22(100%)M ainStrand =Plus /PlusQ uery:1cacaccctcg tg acagg gg g gg 22 ||||||||||||||||||||||Sbjct:1cacaccc tcg tg aca gg gg gg g 22 ........................La mbda K H fina lity1.370.7111.31Gapped..................... 上面简单描述了BLAST 的三个方面的使用方法,当然其中还有很多细节的东西本文没有涉及到,这些细节还需读者在应用BLAST 的过程中慢慢捉摸和体会,同时读者可以参见文献〔4,5,6〕.本文能起到抛砖引玉的作用也就达到了文章的目的.有了本文的介绍,再借助一些帮助文件,相信大家一定能够很快熟悉并掌握BLAS T 的用法,使之成为我们科研工作中方便的工具.参考文献:[1] Altschul S F ,Gish W ,M iller W ,et al .Ba sic loca l alig nm ent sea rch too l [J ].J .Mol .Biol .,1990,215:403~412.[2] Altschul S F ,M adden T L ,Schaffer A A ,et al .Gapped BL A ST and P SI BL AS T :a new ge ner atio n of pro teindatabase search pro g rams [J].N ucl .Acids Res .,1997,25:3389~3402.[3] W o ot to n J C,Federhen S.Statistics of local complex ity in amino acid sequences and sequence databas es [J].Com-puters &Chemistry ,1993,17:149~163.[4] 郝柏林,张淑誉.生物信息学手册[M ].上海:上海科学技术出版社,2000.10,184~210.[5] 赵南明,周海梦.生物物理学[M ].北京:高等教育出版社,2000.7,209~228.[6] 贺林.解码生命—人类基因组计划和后基因组计划[M ].北京:科学出版社,2000.4,421~426.A Brief Introduction of th e Bioinfo rmatics Tool BlastLU Jun 1,3,ZHANG Ying 3,FEN G Li -qing 2,LI Hong1(boratory of The oretical Physics and B iology ,NeiMongol University ,Hohhot 010021,P RC ;2.Department of Physics ,NeiMongol National University ,Tongliao 028043,P RC ;3.Teac hing and Researc h Section of Physics ,N eiMongol Polytechnic University ,Hohhot 010062,P RC ) Abstract :The usag e o f Blast is introduced by o n -line service ,E -mail service and local running respectiv ely ,in o rder to make ev erybo dy master it as soo n as possible,and to make it become a va luable tool in studying theo retical biolog y.Key words :BLAST ;database ;search187第2期吕军等 生物信息学工具BLAS T 的使用简介。
生物信息学课后习题及答案(由10级生技一、二班课代表整理)一、绪论1.你认为,什么是生物信息学?采用信息科学技术,借助数学、生物学的理论、方法,对各种生物信息(包括核酸、蛋白质等)的收集、加工、储存、分析、解释的一门学科。
2.你认为生物信息学有什么用?对你的生活、研究有影响吗?(1)主要用于:在基因组分析方面:生物序列相似性比较及其数据库搜索、基因预测、基因组进化和分子进化、蛋白质结构预测等在医药方面:新药物设计、基因芯片疾病快速诊断、流行病学研究:SARS、人类基因组计划、基因组计划:基因芯片。
(2)指导研究和实验方案,减少操作性实验的量;验证实验结果;为实验结果提供更多的支持数据等材料。
3.人类基因组计划与生物信息学有什么关系?人类基因组计划的实施,促进了测序技术的迅猛发展,从而使实验数据和可利用信息急剧增加,信息的管理和分析成为基因组计划的一项重要的工作。
而这些数据信息的管理、分析、解释和使用促使了生物信息学的产生和迅速发展。
4简述人类基因组研究计划的历程。
通过国际合作,用15年时间(1990-2005)至少投入30亿美元,构建详细的人类基因组遗传图和物理图,确定人类DNA的全部核苷酸序列,定位约10万基因,并对其他生物进行类似研究。
1990,人类基因组计划正式启动。
1996,完成人类基因组计划的遗传作图,启动模式生物基因组计划。
1998完成人类基因组计划的物理作图,开始人类基因组的大规模测序。
Celera公司加入,与公共领域竞争启动水稻基因组计划。
1999,第五届国际公共领域人类基因组测序会议,加快测序速度。
2000,Celera公司宣布完成果蝇基因组测序,国际公共领域宣布完成第一个植物基因组——拟南芥全基因组的测序工作。
2001,人类基因组“中国卷”的绘制工作宣告完成。
2003,中、美、日、德、法、英等6国科学家宣布人类基因组序列图绘制成功,人类基因组计划的.目标全部实现。
2004,人类基因组完成图公布。
一、名词辨析(每题5分,共20分)1、基因与基因组:Gene 基因:遗传功能的单位。
它是一种DNA序列,在有些病毒中则是一种RNA 序列,它编码功能性蛋白质或RNA分子。
Genome 基因组:染色体组,一个生物体、细胞器或病毒的整套基因;例如,细胞核基因组,叶绿体基因组,噬菌体基因组。
2、相似性与同源性:所谓同源序列,简单地说,是指从某一共同祖先经趋异进化而形成的不同序列。
同源性可以用来描述染色体—“同源染色体”、基因—“同源基因”和基因组的一个片断—“同源片断”必须指出,相似性(similarity)和同源性(homology)是两个完全不同的概念。
相似性是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低。
相似性本身的含义,并不要求与进化起源是否同一、与亲缘关系的远近、甚至于结构与功能有什么联系。
3、CDS与cDNA:cDNA序列:互补DNA序列,指的是mRNA为在逆转录酶的作用下将形成DNA 的过程。
CDS序列:编码序列,从起始密码子到终止密码子的所有序列。
4、数据库搜索和数据库查询:数据库查询:对序列、结构以及各种二次数据库中的注释信息进行关键词匹配查找(又称数据库检索)。
数据库搜索:通过特定的序列相似性比对算法,找出核酸或蛋白质序列数据库中与检测序列具有一定程度相似性的序列。
搜索对象不是数据库的注释信息,而是序列信息。
二、判断题(20分)1、生物信息学可以理解为生命科学中的信息科学。
(√)2、DNA分子和蛋白质分子都含有进化信息。
(√)3、目前生命科学研究的重点和突破点的已完全转移到生物信息学上,已不需要实验做支撑。
(×)4、生物信息学的发展大致经历了三个阶段:前基因组时代、基因组时代和后基因组时代。
(√)5、基因组与蛋白质组一样,都处于动态变化之中。
(×)6、蛋白质三维结构都是静态的,在行使功能的过程中其结构不会改变。
(×)7、生物信息学中研究的生物大分子主要是脂类和多糖。
NCBI在线Blast的图文说明Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST 结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP 是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN 是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
NCBI的在线blast:/Blast.cgi1、进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
NCBI在线blast页面2、粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
NCBI在线blast页面3、blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
blast参数设置4、注意一下你输入的序列长度。