NCBIblast使用教程[1]
- 格式:ppt
- 大小:2.30 MB
- 文档页数:76
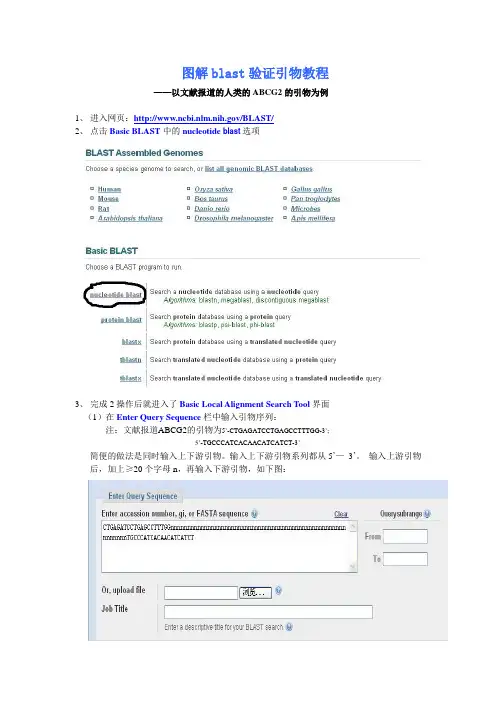
图解blast验证引物教程——以文献报道的人类的ABCG2的引物为例1、进入网页:/BLAST/2、点击Basic BLAST中的nucleotide blast选项3、完成2操作后就进入了Basic Local Alignment Search Tool界面(1)在Enter Query Sequence栏中输入引物序列:注:文献报道ABCG2的引物为5’-CTGAGATCCTGAGCCTTTGG-3’;5’-TGCCCATCACAACATCATCT-3’简便的做法是同时输入上下游引物。
输入上下游引物系列都从5’—3’。
输入上游引物后,加上≥20个字母n,再输入下游引物,如下图:(2)在Choose Search Set栏中:Database根据预操作基因的种属定了,本引物可选Human genomic + transcript或Others (nr etc.)。
本人倾向于选后者,觉得此库信息更多。
如下图:(3)在Program Selection中:选择Somewhat similar sequences (blastn)项,如下图:(4)在此界面最下面:如下图Show results in a new window项是显示界面的形式,可选可不选,在此我们选上了。
关键要点击Algorithm parameters参数设置,进入参数设置界面。
4. 参数设置:(1)在General Parameters中:Expect thresshold期望阈值须改为1000,大于1000也可以;在Word size的下拉框将数字改为7。
如下图:(2)Scoring Parameters无须修改(3)Filters and Masking中,一般来说也没有必要改5.点击最下面一栏的BLAST按钮,如图:6.点击BLAST按钮后,跳转出现如下界面:7. 等待若干秒之后,自动跳转出现显示BLAST结果的网页。
该网页用三种形式来显示blast的结果。

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool )是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST采用一种局部的算法获得两个序列中具有相似性的序列。
Blast 中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLAST)是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLAST是蛋白序列到核酸库中的一种查询。
与BLAST)相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36 种比对阵列。
NCBI的在线BLAST下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast 所有的核酸或蛋白序列。
不同的blast 程序上面已经有了介绍。
这里以常用的核酸库作为例子。
BLAST Assembled GenomesChoose 3 species genome to search, or list all qenoiriic BLAST databases.HumanMouseRatAnafeftfoosfe 册a 帰fffl Otyza sativa口Bg tavTUS D Danic思Mo 应Drosoo打ifa tnElanog^趙F□Basic BLAST以核昔醱库的曰则为例Choose a BLTXST gram torun./ --------- > nucleoliJe blast ■5;Search 3nucleotide database using a nucleotide query AfQorithms. bhstn, megablast, discontiguous megablasttintEm bl可st Search p「otein database using a 卩rateinqueryAfgoriihms' bhstp,卩si-blast,卩hi-blastblastK Search protein database using 玄 translated nucleotide querytbiastn Search transialed nucleotide database using a protein querySearch transiated nucleotide database using a 1ranslat«tl nucleotide query2,粘贴fasta格式的序列。

NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。


NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。


科研实验中的生物信息学工具使用教程生物信息学是将数学、统计学和计算机科学应用于生物学研究的交叉学科。
在现代科研中,生物信息学工具已经成为了生物学实验和研究的重要组成部分。
本文将介绍几种常用的生物信息学工具,并提供详细的使用教程。
1. BLAST(Basic Local Alignment Search Tool)BLAST是生物信息学领域中最常见的工具之一,用于在数据库中快速比较DNA或蛋白质序列的相似性。
以下是使用BLAST进行基本比对的步骤:(1)打开NCBI网站,并进入BLAST页面。
(2)选择“nucleotide”或“protein”,取决于你要比对的序列类型。
(3)复制粘贴或上传你要比对的序列。
(4)选择合适的数据库进行搜索,如“nr”(非冗余数据库)。
(5)点击“BLAST”按钮,等待搜索结果。
BLAST会为你提供一个比对报告,其中包含了与你的查询序列相似的序列列表。
2. EMBOSS(European Molecular Biology Open Software Suite)EMBOSS是一个开源的生物信息学软件包,提供了一系列用于序列分析和比对的工具。
以下是使用EMBOSS进行序列分析的步骤:(1)打开EMBOSS软件(可以下载并安装在你的计算机上)。
(2)选择合适的工具,如“water”(Smith-Waterman比对算法)。
(3)输入查询序列和数据库序列。
(4)设置相关参数,如匹配分数和距离惩罚。
(5)点击“Run”按钮,等待分析结果。
EMBOSS将为你提供一个比对报告,并给出一些统计数据,如匹配分数和最佳比对。
3. R/BioconductorR是一种统计软件和编程语言,Bioconductor是R语言的一个生物信息学扩展包,提供了丰富的生物信息学工具和分析方法。
以下是使用R/Bioconductor进行基因表达分析的步骤:(1)打开R软件并加载Bioconductor包。

NCBI在线BLAST使用方法与结果详解NCBI在线BLAST(Basic Local Alignment Search Tool)是一种广泛使用的生物信息学工具,用于比对和分析DNA、RNA或蛋白质序列。
它可以对已知和未知序列进行,找到与查询序列相似的序列,并提供有关相似性和功能的信息。
使用NCBI在线BLAST可以分为四个主要步骤:选择BLAST程序,输入查询序列,选择目标数据库,解析和分析结果。
第一步:选择BLAST程序NCBI提供了多种BLAST程序可供选择,包括BLASTN(DNA对DNA的比对)、BLASTP(蛋白质对蛋白质的比对)、BLASTX(DNA对蛋白质的比对)等。
根据实际需求选择相应的BLAST程序。
第二步:输入查询序列在查询序列的文本框中输入待比对的序列。
可以输入单个序列,也可以上传包含多个序列的文件。
如果输入的序列是DNA或RNA序列,需要选择相应的序列类型。
此外,还可以选择是否使用掩码序列或低复杂性筛选来优化比对结果。
第三步:选择目标数据库用户可以选择目标数据库来与查询序列相似的序列。
NCBI提供了多个常用的数据库,如nr(非冗余蛋白质数据库)、nt(核酸数据库)等。
此外,还可以选择特定的物种数据库来限制比对范围。
第四步:解析和分析结果在BLAST运行完成后,会生成一个结果页面,其中包含了比对结果的详细信息。
结果页面包括比对统计信息、序列比对图、E值、分数等。
通过分析这些信息,可以了解查询序列与目标数据库中的序列之间的相似性和可能的功能。
此外,NCBI在线BLAST还提供了一些高级选项,例如使用特定的算法或参数来进行比对、设置比对阈值、选择比对输出格式等。
这些选项可以根据实际需求进行调整。
总结起来,使用NCBI在线BLAST可以通过选择BLAST程序、输入查询序列、选择目标数据库以及解析和分析结果来比对和分析序列。
通过权衡算法和参数选择,在特定数据库中找到与查询序列相似的序列,从而获得有关其相似性和功能的信息。

蛋白blast的操作方法
蛋白质BLAST是一种用于比对和识别已知蛋白质序列的工具。
下面是进行蛋白质BLAST的基本操作方法:
1. 打开NCBI(国家医学图书馆)的BLAST页面,网址为
2. 选择适合的蛋白质数据库。
在页面的“blastp”部分,可以选择不同的蛋白质数据库,如“nr”(NCBI非冗余蛋白质数据库),“pdb”(蛋白质数据银行)等。
3. 复制或输入要比对的蛋白质序列。
在页面的“Enter Query Sequence”部分,可以粘贴或输入要比对的蛋白质序列。
4. 设置其他参数。
可以根据需要设置其他参数,如过滤序列中的低复杂性区域,调整比对结果的显示方式等。
5. 点击“BLAST”按钮开始比对。
点击页面底部的“BLAST”按钮,系统将开始进行蛋白质比对。
6. 等待比对结果。
根据输入的蛋白质序列的长度和数据库的大小,比对过程可能需要一段时间。
可以在页面上监视比对进度。
7. 查看比对结果。
比对完成后,系统将显示比对结果。
可以查看比对的蛋白质序列,比对的统计信息和潜在的同源蛋白质。
8. 解释和分析比对结果。
根据比对结果,可以解释蛋白质的结构和功能,进一步分析和研究。
以上是蛋白质BLAST的基本操作方法,根据具体需求和研究目标,还可以进行更多的参数设置和高级分析。


NCBI中Blast种类及使用简介NCBI中Blast种类简介1. Blast Assembled Genomes在一个选择的物种基因组序列中去搜索。
2.Basic Blast2.1 nucleotide blast--- 用核酸序列到核酸数据库中进行搜索,包括3个程序2.1.1 Blastn----核酸序列(n)到核酸序列数据库中搜索,是一种标准的搜索。
2.1.2 megablast----该程序使用“模糊算法”加快了比较速度,可以用于快速比较两大系列序列。
可以用来搜索一匹ESTs序列和大的cDNA或基因组序列, 适用于由于测序或者其他原因形成的轻微的差别的序列之间的比较2.1.3 discontiguous megablast----与megablast不同的是主要用来比较来自不同物种之间的相似性较低的分歧序列。
2.2 Protein Blast2.2.1 Blastp ---蛋白质序列到蛋白质序列数据库中搜索,是一种标准的搜索。
2.2.2 psi-blast---位点特异迭代BLAST —用蛋白查询来搜索蛋白资料库的一个程式。
所有被BLAST发现的统计有效的对齐被总和起来形成一个多次对齐,从这个对齐,一个位置特异的分值矩阵建立起来。
这个矩阵被用来搜索资料库,以找到额外的显著对齐,这个过程可能被反复迭代一直到没有新的对齐可以被发现。
2.2.3 PHI-BLAST---以常规的表达模型为特别位置进行PSI - BLAST检索,找出和待查询序列具有一样的表达模型且具有同源性的蛋白质序列。
2.3 Translating BLAST2.3.1 blastx----先将待查询的核酸序列按6 种读框翻译成蛋白质序列,然后将翻译出的蛋白质序列与NCBI 蛋白质序列数据库比较。
2.3.2 tblastn-----先将核酸序列数据库中的核酸序列按6 种读框翻译成蛋白质序列,然后将待查询的蛋白质序列与翻译结果进行比较。
NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
N C B I在线B L A S T使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、3、4、5、NCBI1blast 2,粘贴blast 3,blast4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
注意分值与E值。
分值越大越靠前了,E值越小也是这样。
7,blast结果的详细比对结果。
注意比对到的序列长度。
评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。
加上长度的话,就有四个标准了。
如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一点。
由Qurey(起始1)和Sbjct(起始35)的起始位置可知,5'端是是多了一段的。
有时也要注意3'端的。
附:来源于网络E值(Expect):表示随机匹配的可能性,E值越大,随机匹配的可能性也越大。
E值接近零或为零时,具本上就是完全匹配了。
一致性(Identities):或相似性。
匹配上的碱基数占总序列长的百分数。
缺失或插入(Gaps):插入或缺失。
用"—"来表示。
来源于网络。
NCBI在线BLAST使用方法与结果详解BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA 数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
Blast中常用的程序介绍:1、BLASTP是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
2、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
3、BLASTN是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
下面是具体操作方法1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。
注意显示的最大的结果数跟E值,E值是比较重要的。
筛选的标准。
最后会说明一下。
4,注意一下你输入的序列长度。
注意一下比对的数据库的说明。
5,blast结果的图形显示。
没啥好说的。
6,blast结果的描述区域。
NCBI在线版Blast使用(超详细奥)首先进行Blast类型的选择:blastp:将待查询的蛋白质序列及其互补序列一起对蛋白质序列数据库进行查询;blastn:将待查询的核酸序列及其互补序列一起对核酸序列数据库进行查询;blastx:先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋白质序列,然后将翻译结果对蛋白质序列数据库进行查询;tblastn:先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后将待查询的蛋白质序列及其互补序列对其翻译结果进行查询;tblastx:•先将待查询的核酸序列和核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后再将两种翻译结果从蛋白质水平进行查询。
基本步骤如下:1,进入在线blast界面,可以选择blast特定的物种(如下)。
不同的blast程序上面已经有了介绍。
这里以常用的Blast 中nucleotide blast作为例子。
•Human 人类•Mouse 小鼠•Rat 大鼠•Arabidopsis thaliana拟南芥•Oryza sativa水稻•Bos taurus牛•Danio rerio斑马鱼•Drosophila melanogaster黑腹果蝇•Gallus gallus乌骨鸡•Pan troglodytes黑猩猩•Microbes微生物•Apis mellifera蜜蜂2,粘贴fasta格式的序列(可以是多条奥!!)或使用Accession number(s)、gi(s)(注意仅使用数字,不加上标志符gi)。
选择一个要比对的数据库,如果是人和鼠则进行相应的选择,否则选择Others中的nr/nt 。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
其他选项不是必选的,如Job Title就是这次比对的名字,随便起一个即可;Organism为物种,可以填入你想比对的物种(分类单元如green plant等)的名字(拉丁名字,输入几个字母后会出现索引的)。
NCBI在线BLAST使用方法与结果详解NCBI(National Center for Biotechnology Information)是一个包含大量基因组学、生物信息学等相关数据和工具的数据库。
其中,BLAST (Basic Local Alignment Search Tool)是一种常用的序列比对工具,可用于在数据库中搜索相似序列。
一、BLAST简介BLAST是一种基于序列比对的方法,可用于确定一给定序列与数据库中序列的相似性。
其工作原理是将查询序列与数据库中的序列进行比对,并生成一个比对得分来衡量它们之间的相似程度。
通过BLAST的结果,可以获得序列的匹配位置、长度、相似性等信息,从而帮助研究人员进行更深入的生物学研究。
二、使用方法1. 打开NCBI网站首先,打开浏览器,输入NCBI的网址(https:///),进入NCBI的官方网站。
2. 进入BLAST页面在NCBI的主页上,找到“BLAST”或“BLAST and Alignments”选项,并点击进入BLAST页面。
3. 输入查询序列在BLAST页面上,找到“Enter Query Sequence”或“Enter accession number, gi, or FASTA sequence”等文本框,将需要查询的序列输入其中。
可以直接复制粘贴序列,或选择上传文件的方式输入。
4. 选择数据库在BLAST页面上,找到“Choose Search Set”或“Database”等选项,选择需要比对的数据库。
NCBI提供了多个数据库,如“nr”(非冗余蛋白数据库)、“nt”(非冗余核酸数据库)等,根据研究需要选择合适的数据库。
5. 设置参数根据需要,可以通过“Algorithm parameters”等选项来设置比对参数,如设置匹配的阈值、比对的方式等。
6. 运行BLAST设置完成后,点击“BLAST”或“Run BLAST”等按钮运行BLAST。
如何使用NCBI中的BlastNCBI(National Center for Biotechnology Information)是一个提供生物信息学数据库和工具的综合性资源平台。
其中,BLAST(Basic Local Alignment Search Tool)是一种经典的序列比对工具,用于比对和分析DNA、RNA和蛋白质序列的相似性。
使用NCBI中的BLAST可以有多种方式,包括在线使用和本地使用。
下面将对这两种使用方式进行详细介绍。
一、在线使用NCBIBLASTNCBI提供了一个在线的BLAST界面,用户可以直接在浏览器中使用。
具体步骤如下:1. 打开NCBI网站,点击"Blast"选项卡,然后选择需要比对的序列类型,例如,DNA、蛋白质或者其他。
2. 复制并粘贴待比对的序列到"Enter Query Sequence"文本框中。
或者,您也可以选择上传一个FASTA格式的文件。
3.选择适当的数据库。
NCBI提供了多个数据库供选择,根据您的研究目的选择合适的数据库。
4.配置其他参数。
您可以选择不同的比对算法、设置匹配参数、设定范围等。
5.点击"BLAST"按钮开始比对。
该过程可能需要一些时间,取决于比对数据的大小和服务器的负载情况。
6.一旦比对完成,系统将生成一个结果页面,显示比对结果。
您可以查看比对的统计信息、序列相似性分析、注释信息等。
7.针对一些结果,您可以选择进一步分析和操作,例如,设计引物、进行序列比对、构建进化树等。
二、本地使用NCBIBLAST3.准备待比对的序列,并保存到FASTA格式的文件中。
4.打开终端或命令提示符,并导航到BLAST软件的安装目录。
5. 运行BLAST命令。
根据您的比对需求,运行适当的BLAST命令,例如,“blastn”用于DNA比对,”blastp”用于蛋白质比对。
6.设置适当的输入参数,包括查询序列文件、目标数据库、比对算法等。