原创:新版GMP现场检查指导原则与新版ISO 13485对比(连载一)
- 格式:docx
- 大小:33.09 KB
- 文档页数:13

版药品GSP现场检查指导原则随着药品市场的不断发展,药品质量安全成为人们关注的热点话题之一。
质量安全管理是制药企业不断发展壮大的关键借口之一,而版药品GSP(Good Supply Practice)现场检查就是质量安全管理的重要内容之一。
为了保障药品的质量安全,灵活使用版药品GSP现场检查指导原则是十分重要的。
首先,版药品GSP现场检查指导原则的基础是对法规和标准的严格遵守。
电子药监1.0版本的发布,为药品的生产、流通及销售开创了科技化管理的先河,也使版药品GSP现场检查迈向数字化时代。
但是就像传统的版药品GSP现场检查一样,对药品的质量等方面还是要遵守国家规定的标准,特别是在药品生产和质量控制方面要严格按照法规进行执行。
同时,要对从事版药品GSP现场检查的人员进行培训和集中力度管理,使得行动更加有组织、有针对性。
其次,要充分利用信息化技术,包括硬件和软件,在药品的生产、流通及销售的整个过程中进行协同管理,并解决不符合要求的问题。
要根据药品的特殊性,建设相应的信息系统和管理平台,以便及时掌握、分析、研判药品流通中出现的问题,并及时开展整改措施。
此外,利用可视化技术对版药品GSP现场检查结果进行直观化展示,使得相关人员能够快速准确地获得数据,从而为及时发现问题奠定基础。
再次,要增强风险意识,针对风险加强版药品GSP现场检查的频率。
对不同的企业和药品种类要进行分类管理和风险评估,并将不同分类的药品质量控制和版本管理纳入不同级别的版药品GSP现场检查计划中。
及时更新相关的漏洞和薄弱环节,进一步提高药品质量和生产效率。
最后,要加强版药品GSP现场检查结果的反馈和总结,实现经验的共享和归纳总结。
要扩大扫描范围,将版药品GSP现场检查的结果和信息公开,推动各个生产企业、销售单位帮助制定改进药品质量管理的办法及措施,全面提高药品质量和安全的保障水平。
总体而言,版药品GSP现场检查指导原则对于保障药品质量安全起到了十分重要的作用。

新版医疗器械GMP现场检查指导原则与ISO 13485:2016差异对比
2014年12月29日,国家食品药品监督管理总局发布了新版医疗器械生产质量管理规范(2014年第64号,以下简称“新版GMP”),其具体实施时间因产品类型和企业开办情况而各异。
另外,2016年3月1日,ISO官网发布了ISO 13485:2016,其具体实施时间有待确定。
大部分的医疗器械企业不但满足于在本国上市产品,而且想走出国门。
为了帮助企业更好地建立医疗器械质量管理体系,同时符合新版GMP和新版ISO 13485的要求,提高医疗器械质量管理效率,整理了新版GMP及其现场检查指导原则(以下简称“检查原则”),与新版ISO 13485的对比,并稍作浅析,供参考。
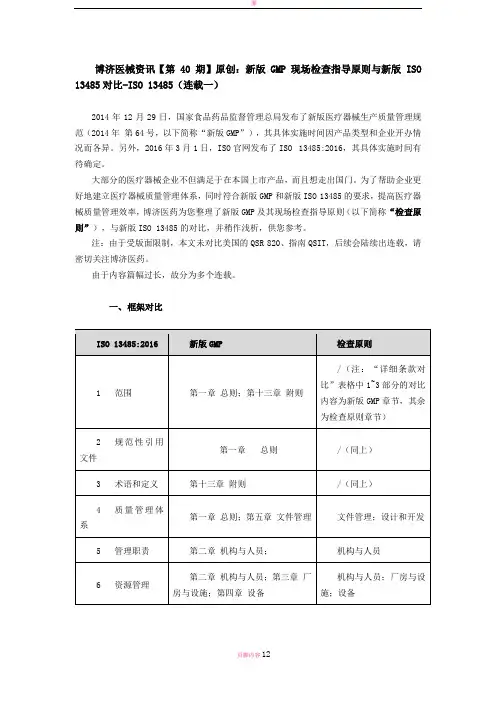
一、框架对比
二、详细条款对比
以ISO 13485:2016为主导。
差异内容标注为不同颜色的加粗字体。

药品生产现场检查风险评定指导原则药品监督管理部门对在企业现场检查中发现的缺陷应根据本指导原则进行分类,附件列举了部分缺陷事例及其分类情况,旨在规范药品检查行为,指导药品检查机构(人员)对发现的缺陷进行科学评定。
本指导原则适用于药品监督管理部门组织的药品GMP认证检查、跟踪检查等检查工作;在药品飞行检查中,涉及药品GMP执行情况的,也可参照本指导原则进行检查和判定。
一、缺陷的分类缺陷分为“严重缺陷”、“主要缺陷”和“一般缺陷”,其风险等级依次降低。
(具体举例见附件1~3)(一)严重缺陷严重缺陷是指与药品GMP要求有严重偏离,产品可能对使用者造成危害的缺陷。
属于下列情形之一的为严重缺陷:1.对使用者造成危害或存在健康风险;2.与药品GMP要求有严重偏离,给产品质量带来严重风险;3.有文件、数据、记录等不真实的欺骗行为;4.存在多项关联主要缺陷,经综合分析表明质量管理体系中某一系统不能有效运行。
(二)主要缺陷主要缺陷是指与药品GMP要求有较大偏离的缺陷。
属于下列情形之一的为主要缺陷:1.与药品GMP要求有较大偏离,给产品质量带来较大风险;2.不能按要求放行产品,或质量受权人不能有效履行其放行职责;3.存在多项关联一般缺陷,经综合分析表明质量管理体系中某一系统不完善。
(三)一般缺陷一般缺陷是指偏离药品GMP要求,但尚未达到严重缺陷和主要缺陷程度的缺陷。
二、产品风险分类企业所生产的药品,依据风险高低分为高风险产品和一般风险产品。
(一)高风险产品以下产品属高风险产品:1.治疗窗窄的药品;2.高活性、高毒性、高致敏性药品(包括微量交叉污染即能引发健康风险的药品,如青霉素类、细胞毒性、性激素类药品);3.无菌药品;4.生物制品(含血液制品);5.生产工艺较难控制的产品(是指参数控制的微小偏差即可造成产品不均一或不符合质量标准的产品,如:脂质体、微球、某些长效或缓释、控释产品等)。
(二)一般风险产品指高风险产品以外的其他产品。

博济医械资讯【第40期】原创:新版GMP现场检查指导原则与新版ISO 13485对比-ISO 13485(连载三)
2014年12月29日,国家食品药品监督管理总局发布了新版医疗器械生产质量管理规范(2014年第64号,以下简称“新版GMP”),其具体实施时间因产品类型和企业开办情况而各异。
另外,2016年3月1日,ISO官网发布了ISO 13485:2016,其具体实施时间有待确定。
大部分的医疗器械企业不但满足于在本国上市产品,而且想走出国门。
为了帮助企业更好地建立医疗器械质量管理体系,同时符合新版GMP和新版ISO 13485的要求,提高医疗器械质量管理效率,博济医药为您整理了新版GMP及其现场检查指导原则(以下简称“检查原则”),与新版ISO 13485的对比,并稍作浅析,供您参考。
注:由于受版面限制,本文未对比美国的QSR 820、指南QSIT,后续会陆续出连载,请密切关注博济医药。
由于内容篇幅过长,故分为多个连载。
一、详细条款对比
以ISO 13485:2016为主导。
差异内容标注为不同颜色的加粗字体。

解读新版GMP现场检查指导原则vs新版ISO 134852014年12月29日,国家食品药品监督管理总局发布了新版医疗器械生产质量管理规范(2014年第64号,以下简称“新版GMP”),其具体实施时间因产品类型和企业开办情况而各异。
另外,2016年3月1日,ISO官网发布了ISO 13485:2016,其具体实施时间有待确定。
大部分的医疗器械企业不但满足于在本国上市产品,而且想走出国门。
为了帮助企业更好地建立医疗器械质量管理体系,同时符合新版GMP和新版ISO 13485的要求,提高医疗器械质量管理效率,博济医药为您整理了新版GMP及其现场检查指导原则(以下简称“检查原则”),与新版ISO 13485的对比,并稍作浅析,供您参考。
一、详细条款对比ISO 13485:2016(第8章节)新版GMP/检查原则差异浅析得到满足。
这种监视和测量应依据所策划的文件化的安排和文件化的程序,在产品实现过程的适当阶段进行。
应保持符合接收准则的证据。
授权放行产品的人员身份应予以记录(见4.2.5)。
适当时,记录应识别用于开展测量活动的测试设备。
只有在已策划的文件化的安排已圆满完成时,才能放行产品和交付服务。
对于植入性医疗器械,组织应记录检验和试验人员的身份。
术要求制定产品的检验规程,并出具相应的检验报告或证书。
查看产品检验规程是否涵盖强制性标准以及经注册或者备案的产品技术要求的性能指标;确认检验记录是否能够证实产品符合要求;查看是否根据检验规程及检验结果出具相应的检验报告或证书。
8.3.2需要常规控制的进货检验、过程检验和成品检验项目原则上不得进行委托检验。
对于检验条件和设备要求较高,确需委托检验的项目,可委托具有资质的机构进行检验,以证明产品符合强制性标准和经注册或者备案的产品技术要求。
*8.4.1每批(台)产品均应当有批检验记录,并满足可追溯要求。
8.4.2检验记录应当包括进货检验、过程检验和成品检验的经注册或者备案的产品技术要求制定产品的检验规程;2)检查原则更加细化了产品的检验要求;3)虽然13485未在此章节强调每批(台)产品均应当有批检验记录,并满足可追溯要求,但在前文第“7.5.1 生产和服务提供的控制”有提及;4)植入性医疗器械的相关要求,需参考《GMP植入性医疗器械现场检查指导原则》。

新版G S P现场检查指导原则-CAL-FENGHAI-(2020YEAR-YICAI)_JINGBIAN药品经营质量管理规范现场检查指导原则说明一、为规范《药品经营质量管理规范》检查工作,确保检查工作质量,根据《药品经营质量管理规范》,制定《药品经营质量管理规范现场检查指导原则》。
二、应当按照本指导原则中包含的检查项目和所对应的附录检查内容,对药品经营企业实施《药品经营质量管理规范》情况进行全面检查。
三、按照本指导原则进行检查过程中,有关检查项目应当同时对照所对应的附录检查内容进行检查。
如果附录检查内容检查中存在任何不符合要求的情形,所对应的检查项目应当判定为不符合要求。
四、本指导原则批发企业检查项目共258项,其中严重缺陷项目(**)6项,主要缺陷项目(*)107项,一般缺陷项目145项。
本指导原则零售企业检查项目共180项,其中严重缺陷项目(**) 4项,主要缺陷项(*)58 项,一般缺陷项118项。
五、药品零售连锁企业总部及配送中心按照药品批发企业检查项目检查,药品零售连锁企业门店按照药品零售企业检查项目检查。
3六、结果判定:注:缺陷项目比例数=对应的缺陷项目中不符合项目数/(对应缺陷项目总数-对应缺陷检查项目合理缺项数)×100%。
4第一部分药品批发企业一、《药品经营质量管理规范》部分56789101112131415161718192021222324二、附录部分(一)冷藏冷冻药品的储存与运输管理2526272829(二)药品经营企业计算机系统3031323334(三)温湿度自动监测353637(四)药品收货与验收3839404142434445(五)验证管理4647484950。

![ISO13485与GMP区别[小编推荐]](https://uimg.taocdn.com/dccc7b3aa200a6c30c22590102020740be1ecd9a.webp)
ISO13485与GMP区别[小编推荐]第一篇:ISO13485与GMP区别[小编推荐]ISO13485,ISO9000及GMP的作用与区别***专业代理医疗器械行业相关认证及资质代办:ISO9000认证,ISO13485认证,医疗器械生产许可证,医疗器械经营许可证,医疗器械产品注册证,医疗器械企业标准编写等。
当前我国对医疗器械生产企业实施医疗器械生产企业质量管理规范(简称医疗器械GMP)。
那么,医疗器械GMP与ISO9000、ISO13485标准有何不同?已通过了ISO9000和ISO13485认证的企业,是否可以不实施医疗器械GMP?ISO9000族标准是质量管理体系标准,规定了质量鉴定管理体系的通用要求,ISO9000族标准适用于各种类型的组织,如:生产企业、服务行业、设计研究部门、管理部门。
由于医疗器械是救死扶伤、防病治病的特殊产品,其质量的基本要求是安全有效,仅按照ISO9000族标准的通用要求来规范医疗器械生产企业是不够的。
为此,ISO发布ISO13485标准对医疗器械生产企业的质量管理体系提出了专用要求。
与此同时,各国政府通过立法进一步加强对医疗器械生产企业的监督管理,确保上市医疗器械的安全有效。
如美国通过实施医疗器械GMP、欧盟也采用欧共体医疗器械指令等法规来对医疗器械生产企业提出法规要求。
因此,我国政府根据我国医疗器械生产企业实际情况,提出法规要求和质量体系要求,制定医疗器械GMP也正是基于以上出发点,以确保医疗器械安全有效,为人民健康安全负责。
实施医疗器械GMP是政府的行为,是强制性的,而医疗器械生产企业实施ISO9001和ISO13485质量管理体系认证是自愿的,两者不能相互代替。
当然,通过ISO9001和ISO13485质量管理体系认证有利于实施医疗器械GMP,但企业实施的医疗器械GMP也不等于通过ISO9001和ISO13485质量管理体系认证,因为两者要求不同。
标准的共同点:ISO9001和ISO13485标准的共同着重点在于“完整的管理体系”,而后者更充分考虑并满足了决大部分,国家食品药品监督管理局对医疗器械管理,相关法规和行业的特殊要求:地位——市场准入的强制性要求f5x中国顾问师网。