人cDNA第一链合成M-MLV酶联免疫分析
- 格式:doc
- 大小:106.50 KB
- 文档页数:3


cDNA 第一链合成试剂盒操作方法1、逆转录反应( 1)在无菌薄壁管中加入以下成份:Total RNA 0.5-1.0 μg(dN) 9 or oligo(dT) 18 100pmol( 2)混匀后, 70℃变性 5min,将薄壁管迅速置于冰上冷却,然后依次加入以下成份:5×M-MLV Buffer 4μldNTPs( 10mM each)RNasin 1μl 10U100mM DTTM-MLV Reverse Transcriptase 2μl 100UDEPC-treated water up to 20 μl( 3)混匀后短暂离心,使用oligo(dT) 18 引物时在42℃,使用随机引物(dN) 9 时在37℃下温育 1 小时。
(4) 94 ℃ 5min 终止反应后,冰上冷却。
(5)合成的 cDNA第一链可直接用于 PCR扩增或进行其它下游操作。
2、 PCR扩增( 1)在 PCR薄壁管中加入以下试剂Synthesized cDNA 2-5 μl10×PCR Buffer 2μlUpper primer 10pmolLower primer 10pmoldNTPs(10mM each)0.5 μlTaq DNA Polymerase 1.5UddH2O up to 20 μl( 2)混匀后短暂离心,然后进行PCR扩增。
94℃预变性 2.5min 。
进入下列30~40 个循环(此扩增条件仅供参考):94℃30s, 60℃ 45s ,72℃ 1min ~3min。
最后 72℃延伸 7min。
注意事项1、确保 RNA完整性及纯度。
RNA质量是决定合成cDNA第一链成功的关键因素,其纯度及完整性可由 OD260/OD280比值和进行琼脂糖凝胶电泳检测。
较纯的 RNA 样品 OD260/OD280比值通常为 1.8-2.0 ,若该比值偏低,应进一步纯化。
真核生物RNA样品电泳图谱 28s 和 18s 的比值约为 2:1 ,否则,表明有 RNA降解。

cdna第一链合成所需要的引物CDNA合成是一种在实验室中合成DNA的技术。
在合成CDNA的过程中,引物起着关键的作用。
引物是一种短链DNA或RNA序列,其序列与目标RNA 的反义序列互补。
通过引物的互补配对,CDNA链可以在反转录过程中产生。
在合成CDNA第一链时,需要使用两种引物,即逆转录引物和OLIGO(dT)引物。
逆转录引物是在典型的CDNA合成反应中使用的第一个引物,它通常由RNA 依赖的DNA聚合酶(例如M-MLV逆转录酶)合成。
逆转录引物的设计应囊括目标RNA的起始位点,以确保所有目标RNA分子都能够转录成CDNA。
OLIGO(dT)引物是用于合成mRNA的CDNA的第二个引物。
它是一种具有多个连续脱氧胸腺嘧啶(dT)的短链DNA序列。
这种引物通过与目标mRNA 的多聚腺嘌呤尾部互补,从而将其用作合成CDNA的起始点。
在实际实验中,通常会使用反转录引物和OlIGO(dT)引物的混合物来合成CDNA的第一链。
这是因为许多mRNA分子的5'端没有腺嘌呤残基,所以OLIGO(dT)引物无法与它们互补。
因此,在使用OLIGO(dT)引物无法反转录mRNA分子的情况下,反转录引物可以用于产生CDNA的第一链。
此外,在CDNA合成的过程中,还需要引物包含其他特定序列,以便于在后续实验中对CDNA进行扩增、检测等操作。
例如,引物的末端通常含有限制酶切位点,以便将CDNA放入载体中。
此外,引物还可以带有一组序列标签,如荧光标签或亲和标签,以便实现CDNA的可视化或纯化。
总结起来,合成CDNA第一链所需的引物包括逆转录引物和OLIGO(dT)引物,它们的序列应与目标RNA互补。
合成CDNA的引物设计应考虑目标RNA的特点,并可能包含特定的序列标签或限制酶切位点。
引物的选择及其设计对于成功合成CDNA的第一链至关重要,因此需要进行仔细的序列分析和合成策略的考虑。

M-MLV逆转录酶说明书【货号】【规格】C28025-01110,000 单位C28025-014 50,000 单位C28025-021 200,000 单位浓度:200U/µl 保存于-20℃(勿除霜)【描述】莫洛尼氏鼠白血病毒逆转录酶(M-MLV RT)能以单链RNA或DNA为模板,在引物的引发下合成与模板互补的DNA链。
本逆转录酶是M-MLV的部分pol基因在大肠杆菌中的表达产物(1-3)。
使用M-MLV RT 可以合成长达7kb的第一链cDNA。
【组分】M-MLV逆转录酶5×第一链合成缓冲液[250mM Tris-HCl(pH8.3,室温),375 mM KCl,15mM MgCl2]0.1M DTT【保存缓冲液】20mM Tris-HCl(pH7.5),100mM NaCl,0.1mM EDTA,1mM DTT,0.01%(v/v)NP-40,50%(v/v)甘油【保存条件】将所有的组分保存于-20℃冰箱(勿除霜)。
【额外的组分】仅在使用前将5×第一链合成缓冲液和0.1M DTT在室温解冻,用完后迅速重新冰冻。
RNaseOUT™重组核酸酶抑制剂(40单位/μl)可从Invitrogen单独订购(货号:10777-019)。
【使用M-MLV逆转录酶进行第一链cDNA的合成】建立20μl 的反应体系可以用于逆转录1ng~5μg总RNA或1~500ng mRNA1.将以下组分加入无核酸酶的微量离心管中1μl oligo(dT)12-18(500μg/ml),或50–250ng随机引物或2pmole基因特异性引物1ng-5μg总RNA或1ng-500ng mRNA1μl 10mM dNTP混合物(dATP,dGTP,dCTP和dTTP均为10mM,pH中性)加入灭菌蒸馏水至12μl2.混合物在65℃加热5分钟后,迅速置于冰上冷却。
短暂离心后,加入以下组分:4μl 5×第一链合成缓冲液2μl 0.1M DTT1μl RNaseOUT™核酸酶抑制剂(40单位/μl)(注意:如果起始RNA的量小于50ng,则必须加入RNaseOUT™)3.在离心管中轻轻将各种成分混合,并在37℃下孵育2分钟。

酶联免疫反应的原理
酶联免疫反应(enzyme-linked immunosorbent assay,ELISA)
是一种常用的免疫学实验技术,用于检测和定量分析目标物(例如蛋白质、抗原或抗体)在样本中的存在和浓度。
酶联免疫反应的原理基于特定抗原与其对应的抗体的高度特异性结合。
典型的酶联免疫反应包括以下步骤:
1. 固定:首先,在固相(例如试管、酶标板等)表面上,直接或间接地固定特定抗原或抗体。
直接固定指将抗原或抗体直接吸附在固相上,而间接固定则需要先加入一种可与抗原或抗体结合的二抗。
2. 绑定:样本中的目标物与固定的抗体或抗原结合形成复合物。
如果样本中含有目标物,则目标物将与固定的抗体结合;如果样本中含有抗体,则抗体将与固定的抗原结合。
3. 洗涤:通过洗涤步骤去除未结合的物质,以减少非特异性背景信号。
4. 信号发生:添加与目标物结合的检测抗体。
检测抗体通常会与目标物不同的部位结合。
这个检测抗体可以是已标记的抗体,其标记物可以是酶(例如辣根过氧化物酶或碱性磷酸酶)等,也可以是免疫荧光、生物素/亲和素等。
5. 洗涤:再次通过洗涤步骤去除未结合的检测抗体。
6. 信号测定:将适合于检测的底物(例如染色底物、荧光底物或发光底物)添加到反应体系中,底物在酶的作用下产生可测量的信号。
酶标板读板仪等设备可测量这种信号的强度。
通过测量光学信号的强度,可以确定目标物在样本中的存在和浓度。
由于酶标记的检测抗体产生的信号可被放大,酶联免疫反应具有高灵敏度和特异性,被广泛用于医学诊断、生物学研究以及药物研发等领域。

RNA得提取与c DNA合成原理与实验方法第一节、概述从貞•核生物得组织或细胞中提取mRN A,通过酶促反应逆转录合成c DNA得第一链与第二链,将双链c DNA与载体连接,然后转化扩增,即可获得cDNA文库,构建得c DNA文库可用于真核生物基因得结构、表达与调控得分析:比较cDNA与相应基因组DNA序列差异可确立内含子存在与了解转录后加工等一系列问题。
总之cDNA得合成与克隆已成为当今真核分子生物学得基本手段。
自70年代中叶首例cDNA克隆问世以来,已发展了许多种提高cDNA合成效率得方法,并大大改进了载体系统,目前cDNA合成试剂已商品化。
cDNA合成及克隆得基本步骤包括用反转录酶合成cDNA第一链,聚合酶合成cDNA第二链,加入合成接头以及将双链DNA 克隆到于适当载体(噬菌体或质粒)。
一、RNA制备模板m RNA得质量直接影响到cDNA合成得效率。
由于mRNA分子得结构特点,容易受RNA酶得攻击反应而降解,加上RNA酶极为稳左且广泛存在,因而在提取过程中要严格防止RNA酶得污染,并设法抑制其活性,这就是本实验成败得关键。
所有得组织中均存在RNA酶.人得皮肤、手指、试剂、容器等均可能被污染,因此全部实验过程中均需戴手套操作并经常更换(使用一次性手套)。
所用得玻璃器皿需巻于干燥烘箱中2 0 0°C烘烤2小时以上。
凡就是不能用高温烘烤得材料如塑料容器等皆可用0、1 %得焦碳酸二乙酯(DEPC)水溶液处理,再用蒸憎水冲净。
DEPC就是RNA酶得化学修饰剂,它U RNA酶得活性基团纟110酸得咪哇环反应而抑制酶活性。
DEPC与氨水溶液混合会产生致癌物,因而使用时需小心。
试验所用试剂也可用DEPC处理,加入DEPC至0、1 %浓度,然后剧烈振荡10分钟,再煮沸15分钟或髙压火菌以消除残存得D EPC.否则DEPC也能与腺噪吟作用而破坏mRN A 活性。
但DEPC能与胺与兢基反应,因而含Tris与DTT得试剂不能用DEPC处理。

RACE技术的原理和操作RACE(Rapid Amplification of cDNA Ends)技术是一种用于扩增cDNA末端序列的方法,广泛应用于克隆和鉴定未知基因的研究中。
下面将详细介绍RACE技术的原理和操作步骤。
一、原理:RACE技术是通过逆转录反应将RNA转录成cDNA,然后利用特殊的引物设计,通过聚合酶链式反应(PCR)进行扩增,最终得到所需的cDNA末端序列。
RACE技术主要包括5'-RACE和3'-RACE两个步骤。
5'-RACE:用于扩增未知序列的5'端操作步骤:1.提取总RNA,并进行逆转录反应,得到第一链cDNA。
2.利用聚dT载体和逆转录酶进行第二链合成。
3.利用内源引物(如具有较高表达的基因的已知序列)和外源引物A 进行PCR扩增。
4. 应用Nested PCR进行第二轮扩增,内嵌引物(nested primer)会在前一轮PCR反应的基础上扩增更长的特异性产物。
5.得到扩增的特异性产物后,纯化PCR产物并进行测序分析,获得5'端的未知序列。
3'-RACE:用于扩增未知序列的3'端操作步骤:1.提取总RNA,并进行反转录反应。
2.利用特异性引物(如基因的已知3'端序列)和逆转录酶进行第一轮PCR扩增。
3. 利用Nested PCR进行第二轮扩增,内嵌引物(nested primer)会在前一轮PCR反应的基础上扩增更长的特异性产物。
4.纯化PCR产物并进行测序分析,获得3'端的未知序列。
二、操作:1. 提取总RNA:一般使用RNAiso Plus试剂或类似试剂从细胞或组织中提取总RNA。
2. 逆转录反应:利用逆转录酶将RNA转录成cDNA。
逆转录酶一般有M-MLV逆转录酶和SuperScript III等。
3.第一链合成:利用聚克隆酶将逆转录的cDNA合成第一链,即得到第一链cDNA。
4.第二链合成:利用特殊的引物和逆转录酶将第一链cDNA合成第二链。

酶联免疫反应的原理和步骤酶联免疫反应是一种常用的实验技术,用于检测和定量分析生物样品中的抗原或抗体。
它基于抗原与抗体之间的特异性结合,通过酶的催化作用使其产生可测量的信号。
本文将介绍酶联免疫反应的原理和步骤。
一、原理酶联免疫反应的原理基于抗原与抗体之间的特异性结合。
抗原是一种能够引起免疫系统产生抗体反应的物质,抗体则是由免疫系统产生的一种蛋白质分子。
在酶联免疫反应中,一种特定的抗体被固定在固相(如微孔板)上,待测样品中的抗原与固相上的抗体结合形成抗原-抗体复合物。
然后,另一种与抗体结合的酶标记抗体被加入,与抗原-抗体复合物结合。
最后,通过添加底物使酶催化产生可测量的信号,从而确定待测样品中的抗原或抗体的数量。
二、步骤1. 准备试剂和设备:包括微孔板、抗体、酶标记抗体、底物、洗涤缓冲液等。
确保试剂和设备的质量和保存条件符合要求。
2. 固相吸附:将特定的抗体溶液加入微孔板孔中,使其在孔壁上吸附。
然后,将孔壁上的未结合抗体洗涤掉,以减少非特异性结合。
3. 样品孔加样:将待测样品加入微孔板中,与固相上的抗体发生特异性结合。
对于检测抗体,待测样品即为抗原;对于检测抗原,待测样品即为抗体。
4. 酶标记抗体加样:将与酶标记抗体结合的抗体加入微孔板中,与抗原-抗体复合物结合。
这种酶标记抗体通常与较早加入的抗体具有不同的特异性。
5. 洗涤:将微孔板中的未结合抗体和其他杂质洗涤掉,以减少非特异性结合。
洗涤缓冲液的成分和洗涤次数应根据实验要求进行调整。
6. 底物加样:将含有底物的溶液加入微孔板中,酶催化底物产生可测量的信号。
底物的选择应根据酶的特性和实验要求进行。
7. 反应停止:通过加入反应停止剂,终止酶催化反应。
反应停止剂的选择应根据底物和酶的特性进行。
8. 信号检测:使用光谱仪、荧光仪或比色计等仪器检测底物催化产生的信号。
信号的强度与待测样品中目标物质的浓度成正比。
9. 数据分析:根据信号的强度和标准曲线,计算出待测样品中目标物质的浓度。

实验六酶联免疫吸附测定法摘要:酶联免疫吸附测定(enzyme-linked immunosorbent assay 简称ELISA)是在免疫酶技术(immunoenzymatic techniques)的基础上发展起来的一种新型的免疫测定技术,具有操作简便、灵敏性高等优点,是生物化学等领域检验的常规测定方法。
本实验采用ELISA间接竞争法对牛奶中鼠抗氯霉素(CAP)进行了检测和分析。
关键词:酶联免疫吸附测定鼠抗氯霉素检测60 年代初期,Averameas 及Ram 等在不破坏酶的催化活性及免疫球蛋白的免疫活性或蛋白质结构的前提下,利用特殊的交联剂将辣根过氧化物酶(HRP)与人血清白蛋白(HSA)连接在一起,后来又用酸性磷酸酯酶(AP)与抗体(Ab)结合,这些结合物统称为酶标记物或酶结合物,被用于抗原或抗体的示踪、定位或定量测定,从此建立了免疫酶技术。
1971 年瑞典的Engvall 和荷兰的Van Weeman 等人使抗体与溴化氰激活的纤维素结合或使抗体吸附于聚苯乙烯试管上制成固相免疫吸附剂(固相载体),再与免疫酶技术结合,建立了酶联免疫吸附测定法,用于检测抗体或抗原。
1974 年Voller 等用聚苯乙烯微量反应板作为吸附载体吸附抗体(抗原),再与相应的酶标记物结合,使ELIS A 操作更方便,易重复,灵敏度可高达ng (10-9g)至pg(10-12g),并且所需仪器设备简单,因而成为生物化学,临床医学检验的常规测定方法之一,原则上ELISA 可用于检测一切抗原、抗体及半抗原,可以直接定量测定体液中的可溶性抗原。
在实际应用方面可作疾病的临床诊断、疾病监察、疾病普查、法医检查、兽医及农业上的植物病害的诊断鉴定等。
因此,它和生物化学、免疫学、微生物学、药理学、流行病学及传染病学等方面密切相关。
ELISA 过程包括抗原(抗体)吸附在固相载体上称为包被,加待测抗体(抗原),再加相应酶标记抗体(抗原),生成抗原(抗体)--待测抗体(抗原)--酶标记抗体的复合物,再与该酶的底物反应生成有色产物。

RNA的提取和cDNA合成原理和实验方法第一节. 概述从真核生物的组织或细胞中提取mRNA,通过酶促反响逆转录合成cDNA的第一链和第二链,将双链cDNA和载体连接,然后转化扩增, 即可获得cDNA文库,构建的cDNA文库可用于真核生物基因的构造、表达和调控的分析;比拟cDNA和相应基因组DNA序列差异可确定含子存在和了解转录后加工等一系列问题。
总之cDNA的合成和克隆已成为当今真核分子生物学的根本手段。
自70年代中叶首例cDNA克隆问世以来,已开展了许多种提高cDNA合成效率的方法,并大大改良了载体系统,目前cDNA合成试剂已商品化。
cDNA合成及克隆的根本步骤包括用反转录酶合成cDNA第一链,聚合酶合成cDNA第二链,参加合成接头以及将双链DNA克隆到于适当载体(噬菌体或质粒)。
一.RNA制备模板mRNA的质量直接影响到cDNA合成的效率。
由于mRNA分子的构造特点,容易受RNA 酶的攻击反响而降解,加上RNA酶极为稳定且广泛存在,因而在提取过程中要严格防止RNA 酶的污染,并设法抑制其活性,这是本实验成败的关键。
所有的组织中均存在RNA酶,人的皮肤、手指、试剂、容器等均可能被污染,因此全部实验过程中均需戴手套操作并经常更换〔使用一次性手套〕。
所用的玻璃器皿需置于枯燥烘箱中200℃烘烤2小时以上。
但凡不能用高温烘烤的材料如塑料容器等皆可用0.1%的焦碳酸二乙酯(DEPC)水溶液处理,再用蒸馏水冲净。
DEPC是RNA酶的化学修饰剂,它和RNA酶的活性基团组氨酸的咪唑环反响而抑制酶活性。
DEPC与氨水溶液混合会产生致癌物,因而使用时需小心。
试验所用试剂也可用DEPC处理,参加DEPC至0.1%浓度,然后剧烈振荡10分钟,再煮沸15分钟或高压灭菌以消除残存的DEPC,否则DEPC也能和腺嘌呤作用而破坏mRNA活性。
但DEPC能与胺和巯基反响,因而含Tris和DTT的试剂不能用DEPC处理。
Tris溶液可用DEPC处理的水配制然后高压灭菌。
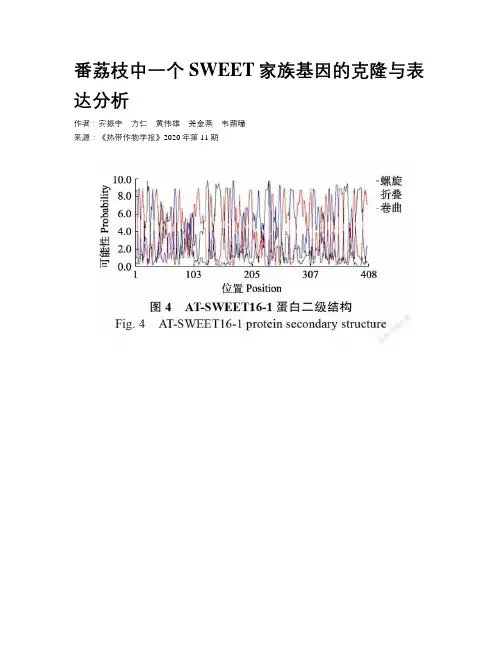
番荔枝中一个SWEET家族基因的克隆与表达分析作者:安振宇方仁黄伟雄尧金燕韦蒴曈来源:《热带作物学报》2020年第11期摘要:以番荔枝不同组织样品为材料,通过基因文库筛选,利用RT-PCR技术克隆出1个1227bp的基因,命名为AT-SWEET16-1,该基因编码408个氨基酸,该氨基酸序列在N端以α螺旋形成THB结构域。
生物信息学分析结果表明,该蛋白分子量为44.8kDa,等电点为8.87。
进化分析结果发现其与海枣(Phoenix dactylifera)相类聚。
qRT-PCR分析结果表明,该基因在植株的根、茎、嫩叶、老叶、花蕾、花苞、幼果、成熟果中均有表达,AT-SWEET16-1基因在不同组织中的表达量依次是:成熟果>茎>根>花蕾>幼果>老叶>嫩叶;该基因在不同果实发育阶段中的表达情况为:在果实不同发育阶段,果柄中的表达量最高,果肉、果皮中则相对较低,种子中最低;但果实成熟期该基因在果柄、果肉中的表达量最高。
原位杂交实验观察发现,基因表达位置为果柄韧皮部、果肉细胞膜间,结合基因表达分析结果,预示该基因在植株糖分积累与转运等方面起到一定的作用。
关键词:番荔枝;克隆;SWEET基因;表达分析中图分类号:S813.3 文献标识码:ACloning and Expression Analysis of a SWEET Family Gene from Annona squamosa L.AN Zhenyu1,2, FANG Ren1*, HUANG Weixiong1, YAO Jinyan1, WEI Shuotong11. Horticultural Research Institute, Guangxi Academy of Agricultural Sciences / Nanning Investigation Station of South Subtropical Fruit Trees, Ministry of Agriculture and Rural Affairs,Nanning, Guangxi 530007, China;2. Guangxi Crop Genetic Improvement and Biotech-Nology Lab, Nanning, Guangxi 530007, ChinaAbstract: The fruit at different stages of Annona squamosa L. was used as the experimental material. A gene named AT-SWEET16-1 with 1227bp was cloned by RT-PCR, which encoding 408 amino acids. The amino acid s equence had a α-helix THB domain at the N-terminal. Bioinformatics analysis showed that the molecular weight of the protein was 44.8kDa, and the isoelectric point was 8.87. Evolutionary analysis showed that it was associated with Phoenix dactylifera). qRT-PCR analysis showed that the gene was expressed in roots, stems, tender leaves, old leaves, flower buds, buds, youngfruitandmaturefruit.TheexpressionofAT-SWEET16-1geneintissuewasasfollows:maturefruit>stem>root>flower bud>young fruit>old leaf>tender leaf. The expression of the gene in different fruit development stages was the highest in fruit stalk, relatively low in pulp and pericarp, and the lowest in seed, and the highest expression in fruit stalk and pulp was at fruit maturity stage. It was found that the gene was expressed in the phloem of the fruit stalk and between the cell membrane of the pulp cell wall, indicating that the gene played a role in the progress of the fruit development and the transport of plant nutrients.Keywords: Annona squamosa L.; cloning; SWEET gene; expression analysisDOI: 10.3969/j.issn.1000-2561.2020.11.001番荔枝(Annona squamosa L.)原产于热带美洲和西印度群岛,现广泛分布于中国台湾、福建、广东、广西、海南和云南等省(区),由于其果实糖分含量较高,每100g果肉组织中的可溶性固形物含量可高达20%,总糖含量达15.3%~18.3%,因此,深受广大甜食爱好者的喜爱。
cDNA合成技术Promega公司的RibocloneR M-MLV(H- ) cDNA合成系统采用M-MLV反转录酶的R Nase H缺失突变株取代AMV反转录酶,使合成的cDNA更长。
该系统的第一链合成使用M-MLV反转录酶,cDNA第二链合成采用置换合成法,采用RNaseH和DNA聚合酶Ⅰ进行置换合成,最后用T4 DNA聚合酶切去单链末端,方法简便易行。
该系统试剂包括:20μg 特异性引物200μl M-MLV第一链缓冲液(5×),配方如下: 250mmol/L Tris·Cl pH8.3(37℃时); 375mm ol/l KCl; 15mmol/L MgCl2; 50mmol/L DTT; 10mmol/L dATP,dCTP,dGTP,dT TP混合物(各2.5mmol/L)2×625μ rRNasinR RNA酶抑制剂10,000μ M-MLV反转录酶,RNase H-5μg 对照RNA400μl M-MLV第二链缓冲液(10×),配方如下:400mmol/L Tris·Cl ,pH7.2; 850mmol/L KCl; 44mmol/L MgCl2;30mmol/L DTT; 0.5mg/ml BSA。
500μ RNase H500μ DNA聚合酶Ⅰ100μ T4 DNA聚合酶Ⅰ2×1.25ml 不含核酸酶的水以上所有试剂除对照RNA需在-70℃保存外,其余均可保存于-20℃,可以合成40μg mRN A。
(一)第一链合成1. 试剂[α-32 P] dCTP (>400Ci/mmol),EDTA(50mM和200mM),TE-饱和酚:氯仿(1:1),7. 5M NH4Ac,乙醇(100%和70%),TE缓冲液。
2. 操作步骤(1) 取一灭菌的无RNA酶的eppendorf管,加入RNA模板和适当引物,每μg RNA使用0. 5μg引物(如使用NotⅠ引物接头,使用0.3μg),用H2 O调整体积至15μl,70℃处理5分钟,冷却至室温,离心使溶液集中在管底,再依次加入。
常用于合成cdna第二链的酶合成cDNA第二链的酶是一种重要的酶类,它在分子生物学和基因工程领域中得到了广泛应用。
cDNA是由RNA反转录成的DNA,它是研究基因表达和功能的重要工具。
cDNA第二链合成酶是一种能够将单链cDNA模板转化为双链cDNA的酶,它是cDNA合成的关键步骤之一。
本文将介绍常用于合成cDNA 第二链的酶的种类、特点、应用及优缺点。
一、常用于合成cDNA第二链的酶种类1. M-MLV逆转录酶M-MLV逆转录酶是一种来源于Moloney Murine Leukemia Virus的逆转录酶,它是一种RNA依赖性DNA聚合酶。
M-MLV逆转录酶具有高度的反转录活性和高度的特异性,能够将RNA模板转化为cDNA。
M-MLV逆转录酶常用于合成cDNA第一链和第二链,它的优点是反转录效率高、特异性好、反转录产物稳定。
但是,M-MLV逆转录酶的缺点是不能够合成全长cDNA,因为它不能够通过RNA结构和RNA序列的复杂性。
2. AMV逆转录酶AMV逆转录酶是一种来源于Avian Myeloblastosis Virus的逆转录酶,它是一种RNA依赖性DNA聚合酶。
AMV逆转录酶具有高度的反转录活性和高度的特异性,能够将RNA模板转化为cDNA。
AMV逆转录酶常用于合成cDNA第一链和第二链,它的优点是反转录效率高、特异性好、反转录产物稳定。
但是,AMV逆转录酶的缺点是不能够合成全长cDNA,因为它不能够通过RNA结构和RNA序列的复杂性。
3. SuperScript III逆转录酶SuperScript III逆转录酶是一种来源于Thermus thermophilus的逆转录酶,它是一种RNA依赖性DNA聚合酶。
SuperScript III逆转录酶具有高度的反转录活性和高度的特异性,能够将RNA模板转化为cDNA。
SuperScript III逆转录酶常用于合成cDNA第一链和第二链,它的优点是反转录效率高、特异性好、反转录产物稳定。
一种构建全长cDNA文库的方法刘红军;李青旺;吴淑云;韩增胜;刘智华;李文烨【期刊名称】《安徽农业科学》【年(卷),期】2007(035)005【摘要】建立了一种以LD-PCR为基础的cDNA文库的快速构建方法.以人胎盘组织为材料获得总RNA,利用引物F1、R2在逆转录酶M-MLV的作用下合成cDNA 第1链,进而利用引物F3、R4在DNA聚合酶的作用下通过LD-PCR方法合成cDNA第2链;双链cDNA经SfiⅠ酶切,通过T4 DNA连接酶连接到经相同酶切的JG45质粒载体后构建成cDNA文库,并对文库的容量、重组率以及多样性进行了分析.结果表明,通过该方法构建的cDNA文库容量约为7.01×105 个/μgds-cDNA,重组率为96%;对随机提取的100个克隆质粒的插入序列进行分析,共获得80个不同的cDNA序列,且未见重复序列.该法构建的cDNA质粒文库具有快速、简单的特点,构建的文库质量符合要求,可用于大规模的基因分析.【总页数】3页(P1305-1306,1316)【作者】刘红军;李青旺;吴淑云;韩增胜;刘智华;李文烨【作者单位】西北农林科技大学动物科技学院,陕西杨凌,712100;西北农林科技大学动物科技学院,陕西杨凌,712100;燕山大学,环境与化学工程学院生物工程系,河北秦皇岛,066004;西北农林科技大学动物科技学院,陕西杨凌,712100;西北农林科技大学动物科技学院,陕西杨凌,712100;西北农林科技大学动物科技学院,陕西杨凌,712100;西北农林科技大学动物科技学院,陕西杨凌,712100【正文语种】中文【中图分类】Q501【相关文献】1.适合于全长cDNA文库构建的猪苓菌核及菌丝体总RNA提取方法比较 [J], 李杨;李海娜;夏颖;刘忻壑;李艳茹;许广波2.全长cDNA文库及构建方法与应用进展 [J], 韩慧霞3.全长cDNA文库构建方法及应用研究 [J], 朱利军;长孙东亭;罗素兰4.几种全长cDNA文库构建方法比较 [J], 毛新国;景蕊莲;孔秀英;赵光耀;贾继增5.一种获得大片段克隆的SMART全长cDNA文库构建方法 [J], 董志敏;李英慧;张宝石;关荣霞;常汝镇;邱丽娟因版权原因,仅展示原文概要,查看原文内容请购买。
PCR常见问题分析与对策PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。
假阴性,不出现扩增条带PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及,④PCR循环条件。
寻找原因亦应针对上述环节进行分析研究。
模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。
⑤模板核酸变性不彻底。
在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应固定不宜随意更改。
酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。
需注意的是有时忘加Taq酶或溴乙锭。
引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。
有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称扩增,对策为:①选定一个好的引物合成单位。
②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应和引物合成单位协商解决。
如一条引物亮度高,一条亮度低,在稀释引物时要平衡其浓度。
③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。
④引物设计不合理,如引物长度不够,引物之间形成二聚体等。
Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。
或100ul,应用多大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul后,再做大体积时,一定要模索条件,否则容易失败。
人cDNA第一链合成(M-MLV)酶联免疫分析
试剂盒使用说明书
本试剂盒仅供研究使用。
检测范围:96T
20pg/ml-480pg/ml
使用目的:
本试剂盒用于测定人血清、血浆及相关液体样本中cDNA第一链合成(M-MLV)含量。
实验原理
本试剂盒应用双抗体夹心法测定标本中人cDNA第一链合成(M-MLV)水平。
用纯化的人cDNA第一链合成(M-MLV)抗体包被微孔板,制成固相抗体,往包被单抗的微孔中依次加入cDNA第一链合成(M-MLV),再与HRP标记的cDNA第一链合成(M-MLV)抗体结合,形成抗体-抗原-酶标抗体复合物,经过彻底洗涤后加底物TMB显色。
TMB在HRP酶的催化下转化成蓝色,并在酸的作用下转化成最终的黄色。
颜色的深浅和样品中的cDNA第一链合成(M-MLV)呈正相关。
用酶标仪在450nm波长下测定吸光度(OD值),通过标准曲线计算样品中人cDNA第一链合成(M-MLV)浓度。
1.标本采集后尽早进行提取,提取按相关文献进行,提取后应尽快进行实验。
若不能马上进行试验,可将标本放于-20℃保存,但应避免反复冻融
2.不能检测含NaN3的样品,因NaN3抑制辣根过氧化物酶的(HRP)活性。
操作步骤
1.标准品的稀释:本试剂盒提供原倍标准品一支,用户可按照下列图表在小试管中进行稀
释。
2.加样:分别设空白孔(空白对照孔不加样品及酶标试剂,其余各步操作相同)、标准孔、
待测样品孔。
在酶标包被板上标准品准确加样50μl,待测样品孔中先加样品稀释液40μl,然后再加待测样品10μl(样品最终稀释度为5倍)。
加样将样品加于酶标板孔底部,尽量不触及孔壁,轻轻晃动混匀。
3.温育:用封板膜封板后置37℃温育30分钟。
4.配液:将30倍浓缩洗涤液用蒸馏水30倍稀释后备用
5.洗涤:小心揭掉封板膜,弃去液体,甩干,每孔加满洗涤液,静置30秒后弃去,如此
重复5次,拍干。
6.加酶:每孔加入酶标试剂50μl,空白孔除外。
7.温育:操作同3。
8.洗涤:操作同5。
9.显色:每孔先加入显色剂A50μl,再加入显色剂B50μl,轻轻震荡混匀,37℃避光显色
10分钟.
10.终止:每孔加终止液50μl,终止反应(此时蓝色立转黄色)。
11.测定:以空白空调零,450nm波长依序测量各孔的吸光度(OD值)。
测定应在加终止
液后15分钟以内进行。
操作程序总结:
计算
以标准物的浓度为横坐标,OD值为纵坐标,在坐标纸上绘出标准曲线,根据样品的OD值由标准曲线查出相应的浓度;再乘以稀释倍数;或用标准物的浓度与OD值计算出标
准曲线的直线回归方程式,将样品的OD值代入方程式,计算出样品浓度,再乘以稀释倍数,即为样品的实际浓度。
注意事项
1.试剂盒从冷藏环境中取出应在室温平衡15-30分钟后方可使用,酶标包被板开封后如未用完,板条应装入密封袋中保存。
2.浓洗涤液可能会有结晶析出,稀释时可在水浴中加温助溶,洗涤时不影响结果。
3.各步加样均应使用加样器,并经常校对其准确性,以避免试验误差。
一次加样时间最好控制在5分钟内,如标本数量多,推荐使用排枪加样。
4.请每次测定的同时做标准曲线,最好做复孔。
如标本中待测物质含量过高(样本OD值大于标准品孔第一孔的OD值),请先用样品稀释液稀释一定倍数(n倍)后再测定,计算时请最后乘以总稀释倍数(×n×5)。
5.封板膜只限一次性使用,以避免交叉污染。
6.底物请避光保存。
7.严格按照说明书的操作进行,试验结果判定必须以酶标仪读数为准.
8.所有样品,洗涤液和各种废弃物都应按传染物处理。
9.本试剂不同批号组分不得混用。
保存条件及有效期
1.试剂盒保存:;2-8℃。
2.有效期:6个月。