羟醛缩合
- 格式:doc
- 大小:482.50 KB
- 文档页数:68


发生羟醛缩合反应的条件
羟醛缩合反应发生的条件主要包括:
1.存在羟醛化合物:羟醛缩合反应是由两个羟醛分子发生缩合反应而成的,因此必须要有羟醛化合物存在。
2.酸催化或碱催化:羟醛缩合反应可以通过酸或碱催化,使得反应速率得到加快,反应剂的物质量明显减少。
3.一定的温度和压力:羟醛缩合反应发生的温度一般在50℃以上,压力一般较低,一般在常压下即可。
4.适当的反应时间:羟醛缩合反应需要合适的反应时间,过长或过短都会影响反应的产率。
5.去除反应产物:随着反应的进行,羟醛缩合产物会逐渐生成,必须通过蒸馏、萃取等方法将其分离和去除,以促使反应向前推进。


羟醛缩合机理一、引言羟醛缩合是有机化学中一种重要的反应类型,也是生物大分子合成中最基本的反应之一。
本文将详细介绍羟醛缩合的机理。
二、羟醛缩合概述羟醛缩合是指在弱碱性条件下,甲醛或其他羰基化合物与具有活泼亲核性能的化合物(如胺、酚等)进行缩合反应,生成稳定的亚甲基或烷基化合物的过程。
在此过程中,甲醛分子首先被氢氧根离子(OH-)攻击,形成甲氧根离子和一个极性质子,然后极性质子进一步被亲核试剂(如胺、酚等)攻击,形成新的碳-碳键。
三、羟醛缩合机理1. 羟醛生成首先,在弱碱性条件下,甲醛分子与氢氧根离子发生反应,生成羟甲基或甲氧根离子。
这个步骤是整个反应过程中最重要的步骤之一。
2. 美拉德反应接着,在第一个步骤的基础上,羟甲基或甲氧根离子与亲核试剂(如胺、酚等)发生缩合反应,形成亚甲基或烷基化合物。
这个步骤被称为美拉德反应。
3. 羟醛缩合的条件羟醛缩合反应需要一定的条件,包括弱碱性环境、适当的温度和压力等。
在实际应用中,常用的催化剂包括氢氧化钠、氢氧化钾和三乙胺等。
四、羟醛缩合反应的机理解析1. 羰基亲核加成羰基亲核加成是指亲核试剂(如胺、酚等)中的负电子对攻击羰基碳上电子不足原子所形成的中间体。
在羟醛缩合反应中,亲核试剂(如胺、酚等)中的负电子对攻击了甲醛分子上电子不足原子所形成的中间体。
2. 产物稳定性分析产物稳定性是指产物分子内部键能与外界环境作用力之间平衡状态下所达到的最低自由能状态。
在羟醛缩合反应中,产物的稳定性是由产物分子内部键能和外界环境作用力之间的平衡状态所决定的。
3. 美拉德反应机理美拉德反应机理是指亲核试剂与甲醛分子发生缩合反应时,中间体的生成和消失过程。
在羟醛缩合反应中,美拉德反应机理包括亲核试剂攻击甲醛分子、中间体生成、中间体消失和产物生成等步骤。
五、羟醛缩合反应的影响因素1. 催化剂种类和用量催化剂种类和用量对羟醛缩合反应有重要影响。
常用的催化剂包括氢氧化钠、氢氧化钾和三乙胺等。



丙醛的羟醛缩合反应方程式丙醛是一种重要的有机化合物,常用于制药和农药工业中。
它是一种醛类化合物,分子式为CH3CHO,具有强烈的刺激性气味。
在化学反应中,丙醛可以发生羟醛缩合反应,生成2,4-二甲基-3-戊酮。
羟醛缩合反应是一种常见的有机反应,也称为肉桂酸合成反应。
该反应是一种加成-消除反应,通过将醛和羟醛反应形成α, β-不饱和羰基化合物来进行。
丙醛的羟醛缩合反应方程式如下所示:
CH3CHO + CH3OH → CH3CHOHCH(OMe)CH3
该反应需要催化剂存在,一般使用碱金属醇盐或稀酸催化剂。
反应条件通常在室温下进行,反应时间为几小时。
此时,丙醛和甲醇反应生成羟乙醛,然后再通过加成-消除反应生成2,4-二甲基-3-戊酮。
羟醛缩合反应可以合成许多不同的化合物,如肉桂酸、香兰素、黄酮类化合物等。
它是一种重要的有机合成反应,被广泛应用于制药和农药工业中。
在实际应用中,丙醛的羟醛缩合反应需要注意反应条件的选择和催化剂的选择。
此外,反应过程中需要保持反应体系的纯净和稳定,以确保产物的纯度和收率。
总之,丙醛的羟醛缩合反应是一种重要的有机合成反应,具有广泛的应用前景。
我们需要不断地深入研究和探索,在实践中不断改进反应条件和催化剂,为化学工业和生物医药行业做出更大的贡献。

羟醛缩合产物羟醛缩合反应是一种重要的有机合成反应,通过该反应可以合成各种有机化合物,包括糖类、氨基酸、杀菌剂等。
羟醛缩合的产物很多,下面将介绍几种常见的羟醛缩合反应以及产物。
1. 阿尔多缩合反应阿尔多缩合反应是一种有机合成中常用的反应之一,通过该反应可以合成糖类和多羟基化合物。
这种反应通常以糖类为底物,羟醛以及碱作为催化剂。
反应过程中,羟醛与碱发生亲核进攻,形成的中间体经过消旋,然后再水解生成目标产物。
阿尔多缩合反应常用于制备各种糖类,如葡萄糖、木糖、纤维素等。
2. 缩醛反应缩醛反应是将羟醛与羟酮缩合生成α,β-不饱和羰基化合物的反应。
该反应常用稀碱溶液作为催化剂,生成的产物具有独特的结构和化学性质。
这种反应可以应用于合成有机化合物,如合成檀香醇、柠檬醛等。
3. 酰胺缩合反应酰胺缩合反应是将羟醛与胺缩合生成酰胺的反应。
该反应通常使用酸性条件催化,产物具有酰胺的结构。
酰胺缩合反应是一种常用的合成肽类化合物的方法,通过合理选择底物和反应条件,可以高效合成肽链。
4. 斯奈普-斯莫夫反应斯奈普-斯莫夫反应是将羟醛与胺缩合生成醚的反应。
该反应以胺为氨源,通过氧化羟醛生成羟基胺中间体,然后再与羟醛发生缩合反应,生成醚化合物。
该反应常用于合成含氮有机化合物,具有重要的应用价值。
总结:羟醛缩合反应是合成有机化合物的重要反应之一,通过选择不同的底物和反应条件可以合成各种有机化合物。
在实际应用中,我们需要根据具体的反应需求选择适当的缩合反应方法,并通过合理设计实验条件和催化剂选择来提高反应的效率和产物的选择性。
这些常见的羟醛缩合反应不仅可以应用于实验室合成,还可以应用于工业生产中,有着广泛的应用前景。

羟醛缩合反应举例一、羟醛缩合反应的基本概念羟醛缩合反应是有机化学中的一种重要反应,也称为亚甲基化反应或醛缩合反应。
它是一种通过羟醛分子与另一有机物分子发生缩合反应形成新的分子结构的化学反应。
在该反应中,羟醛分子(如甲醛、乙醛等)与另一有机物分子(如胺、酚、二元酸等)进行缩合,生成新的分子结构。
二、羟醛缩合反应的机理在羟醛缩合反应中,首先是羟基与甲基之间进行亲核加成,生成一个稳定的中间体。
然后在碱催化下进行去质子化作用,使得羧基脱离产生出水分子,最终形成新的含有C-C键和C-O键的产物。
三、举例:甲醛和苯胺的缩合反应1. 实验原理本实验通过将甲醛和苯胺进行加热并加入催化剂后进行缩合反应,生成2-氨基苯甲酸。
2. 实验步骤(1)将苯胺(1.4g)和甲醛(1.2g)加入到干净的圆底烧瓶中。
(2)加入5ml浓氢氧化钠溶液作为催化剂。
(3)将烧瓶放置在油浴中,加热至沸腾,并继续加热反应30分钟。
(4)取出反应液,加入适量的盐酸进行酸化处理。
(5)用水洗涤产物,过滤后干燥即可得到产物。
3. 实验结果产物经过红外光谱分析,得到了以下结果:主峰出现在1700cm^-1处,表明有一个羧基;主峰出现在1600cm^-1处,表明有一个芳香环;主峰出现在1400-1500cm^-1处,表明有一个胺基;主峰出现在1200-1300cm^-1处,表明有一个C-O键。
通过这些结果可以确定产物为2-氨基苯甲酸。
四、总结羟醛缩合反应是一种非常重要的有机化学反应,在实际生产和科学研究中都有广泛的应用。
本实验以甲醛和苯胺的缩合反应为例,说明了羟醛缩合反应的基本概念、机理和实验操作过程。
通过实验可以得到产物,进一步证明了羟醛缩合反应的可行性和重要性。

羟醛缩合键断裂
【原创实用版】
目录
1.羟醛缩合反应概述
2.羟醛缩合键的断裂原因
3.断裂羟醛缩合键的方法
4.羟醛缩合反应在有机合成中的应用
正文
一、羟醛缩合反应概述
羟醛缩合反应是一种常见的有机化学反应,指的是在催化剂的作用下,醇和羧酸之间形成一个新的化学键——羟醛缩合键。
这种反应广泛应用于有机合成领域,如制药、材料科学等。
二、羟醛缩合键的断裂原因
在有机化合物中,羟醛缩合键具有一定的稳定性,但在特定条件下,这种键可能会发生断裂。
断裂原因主要有以下几点:
1.酸碱催化:当环境中存在强酸或强碱时,羟醛缩合键可能受到攻击,从而导致键的断裂。
2.氧化还原反应:在某些氧化还原反应中,羟醛缩合键也可能被破坏。
例如,某些氧化剂可以与羟醛缩合键发生反应,使其断裂。
3.亲核取代反应:在一些亲核取代反应中,亲核试剂可能攻击羟醛缩合键,导致其断裂。
三、断裂羟醛缩合键的方法
要断裂羟醛缩合键,可以采用以下几种方法:
1.酸碱催化:使用强酸或强碱作为催化剂,在特定条件下使羟醛缩合
键断裂。
2.氧化还原反应:利用氧化剂或还原剂进行反应,使羟醛缩合键断裂。
3.亲核取代反应:使用适当的亲核试剂进行反应,攻击羟醛缩合键,导致其断裂。
四、羟醛缩合反应在有机合成中的应用
羟醛缩合反应在有机合成中具有重要意义,可用于合成各种化合物,如醇、羧酸等。
此外,羟醛缩合反应还可用于制药等领域,如合成抗生素、抗病毒药物等。
羟醛缩合是一种有机反应:烯醇或烯醇负离子和羰基化合物反应形成β-羟基醛或者β-羟基酮,然后发生脱水得到共轭烯酮。
羟醛缩合在有机合成当中很重要,它是形成碳碳单键的关键条件之一,罗宾逊成环反应中有一步就是羟醛缩合反应。
羟醛缩合在大学有机化学课程中常作为一个经典构建碳键的反应进行讲解,并用该反应介绍反应机理。
在普通的羟醛缩合反应中,包涵了酮的烯醇对于醛的亲核加成,形成β-羟基酮或者“羟醛”(广泛出现于各种天然产物及药物中的一种结构单元)。
羟醛缩合在生物化学中也同样广泛存在。
羟醛反应自身由醛缩酶催化,然而该反应不是正式的缩合反应,这是因为过程中并未脱除小分子。
反应在醛和酮之间发生(交叉羟醛缩合),或者在两个醛之间发生,则称为Claisen-Schmidt缩合反应。
这些反应都被冠以发现人的名字莱纳·路德维希·克莱森和J.G.施密特。
他们分别于1880和1881年发表了自己在该领域的论文。
机理:
反应的第一步为羟醛反应,第二步为脱水反应即消除反应。
当分子内有活性羧基的情况下,该脱水反应还会伴随脱羧反应。
羟醛加成产物可通过两种机理进行脱水反应:强碱如:叔丁醇钾、氢氧化钾或氢氧化钠通过烯醇机理进行反应,[10]或通过酸-催化进行的烯醇机理进行反应。
酸催化的羟醛反应机理
酸催化的脱水反应
做碱)
碱催化的羟醛反应 (图例使用−OCH
3
碱催化的脱水反应 (这里通常被错写为简单一步,见E1cB消除反应)。
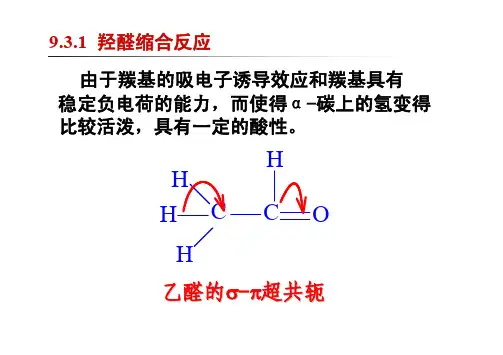
9.3.3.3 羟醛缩合反应
羟醛缩合反应(Aldol c ondensation r eaction)
在稀碱溶液中,两分子有α-H的醛结合生成β-羟基醛的反应。
α-碳上有氢原子的β-羟基醛受热容易失去一分子水,生成α,β-不饱和醛。
反应机理:
β-‐羟基醛
2-丁烯醛(巴豆醛) β-羟基醛在加热时容易脱水生成α,β-不饱和醛。
庚醛以上的醛在碱性溶液中只能得到α,β-不饱和醛,因为分子量大的醛的Aldol反应必须加热才能进行。
easy difficult
酮的羟醛缩合反应不易进行,需要将产物不断
移
出平衡体系,促进反应向右
进行。
如何实现酮的羟醛缩合反应?
Soxhlet Extractor
catalyst
acetone
分子内缩合反应的选择性
交叉的羟醛缩合反应(Crossed
Aldol R eaction)
产物复杂,无实用价值
策略一:其中一个醛无 α-H
Claisen-Schmidt (克莱森-施密特)缩合反应 交叉的羟醛缩合反应(Crossed Aldol R
eaction)
交叉的羟醛缩合反应(Crossed
Aldol R eaction)
策略二:利用酮不易发生羟醛缩合反应的性质
LDA
交叉的羟醛缩合反应(Crossed A ldol R eaction)
Aldol reaction Cannizzaro reaction 三羟甲基乙醛
季戊四醇 。
羟醛缩合键断裂摘要:1.羟醛缩合键的基本概念2.羟醛缩合反应的机理3.羟醛缩合键断裂的原因4.羟醛缩合键断裂的影响5.应对羟醛缩合键断裂的策略正文:羟醛缩合键是一种有机化学中的重要反应,广泛应用于合成复杂有机化合物。
但在实际操作过程中,羟醛缩合键可能会出现断裂现象。
本文将对羟醛缩合键断裂进行分析,探讨其原因、影响及应对策略。
一、羟醛缩合键的基本概念羟醛缩合反应是指两个羟醛分子通过脱水缩合形成一个新的化合物,同时生成一个水分子。
这个过程中,羟醛分子间的羟基和醛基通过缩合形成一个新的化学键,即羟醛缩合键。
二、羟醛缩合反应的机理羟醛缩合反应通常经历以下步骤:首先是醛基的亲核加成,形成一个亚胺中间体;接着亚胺中间体水解,生成一个醇和一个酸;最后,酸与另一个醇发生酯化反应,形成一个新的酯。
在这个过程中,羟醛缩合键逐渐形成。
三、羟醛缩合键断裂的原因1.反应条件过于强烈:在高温、高压或强酸条件下,羟醛缩合反应可能会发生断裂,导致产物失控。
2.溶剂影响:某些溶剂可能会与羟醛分子发生竞争反应,导致羟醛缩合反应受阻,进而引发键断裂。
3.反应物浓度不适:反应物浓度过高或过低都可能导致羟醛缩合反应不完全,从而引发键断裂。
四、羟醛缩合键断裂的影响羟醛缩合键断裂会影响反应的产率和品质。
反应失控可能导致副产物生成,降低目标产物的纯度。
此外,断裂的羟醛缩合键还可能引发其他不良反应,进一步影响产物的结构与性能。
五、应对羟醛缩合键断裂的策略1.优化反应条件:适当调整温度、压力和酸度,以保证羟醛缩合反应的顺利进行。
2.选择适宜的溶剂:通过筛选不同溶剂,找到对羟醛缩合反应有促进作用的溶剂。
3.控制反应物浓度:通过监测反应进程,适时调整反应物浓度,确保反应在适宜范围内进行。
4.引入催化剂:选用合适的催化剂,可以提高羟醛缩合反应的速率和选择性,减少键断裂的发生。
总之,羟醛缩合键断裂是羟醛缩合反应中常见的问题。
羟醛的缩合-概述说明以及解释1.引言1.1 概述羟醛的缩合是一种重要的化学反应,广泛应用于有机合成领域。
羟醛(也称为醛酮)是一类化合物,含有一个碳氧双键和一个碳氢双键的官能团,具有很强的活性。
羟醛缩合指的是两个或多个羟醛分子之间发生的反应,生成一个更大的分子,并且伴随着碳-碳键的形成。
羟醛缩合反应的机理一般包括羟基的负离子产生、亲核试剂的加成和负离子的消除等步骤。
在缩合反应中,羟醛分子发生自身的缩合,可以通过内部的亲核试剂(如羟基)攻击羰基碳上的电子云,从而形成新的碳-碳键。
羟醛缩合在有机合成中具有广泛的应用。
首先,它可以用于构建碳骨架,生成各种有机化合物,如酮、糖和天然产物等。
其次,羟醛缩合反应还可用于合成杂环化合物,如吡咯、噻吩和咪唑等。
此外,羟醛缩合还可以被用作多步合成中的关键步骤,实现合成目标化合物的合成。
综上所述,羟醛缩合反应在有机化学合成中起着重要作用,具有广泛的应用前景。
对羟醛缩合反应的研究不仅有助于深入理解其反应机理,还为合成新型有机化合物提供了有力的工具。
随着合成化学的不断发展,相信羟醛缩合反应将在未来的有机合成中发挥更大的作用。
1.2文章结构1.2 文章结构本文主要介绍了羟醛的缩合反应。
文章结构如下:1. 引言1.1 概述:简要介绍了羟醛的基本概念和缩合反应的背景。
1.2 文章结构:介绍了本文的整体结构和各个章节的内容。
1.3 目的:明确了本文的目标和意义。
2. 正文2.1 羟醛的定义和性质:详细介绍了羟醛的化学结构、命名规则和一些基本性质。
2.2 羟醛缩合反应的机理:探讨了羟醛缩合反应的基本原理和常见的机理路径。
2.3 羟醛缩合在有机合成中的应用:列举了羟醛缩合在有机合成中的一些重要应用,并说明其在合成中的优势和局限性。
3. 结论3.1 总结羟醛缩合的重要性:总结了羟醛缩合在有机合成中的重要性和广泛应用的现状。
3.2 展望羟醛缩合的未来发展:对羟醛缩合反应的未来发展方向和可能的改进进行了展望。
碱催化羟醛缩合反应机理
羟醛缩合反应机理是,具有α-H的醛或酮,在碱催化下生成碳负离子,然后碳负离子作为亲核试剂对醛或酮进行亲核加成,生成β-羟基醛,β-羟基醛受热脱水生成α-β不饱和醛或酮。
在稀碱或稀酸的作用下,两分子的醛或酮可以互相作用,其中一个醛(或酮)分子中的α-氢加到另一个醛(或酮)分子的羰基氧原子上,其余部分加到羰基碳原子上,生成一分子β-羟基醛或一分子β-羟基酮。
这个反应叫做羟醛缩合或醇醛缩合。
通过醇醛缩合,可以在分子中形成新的碳碳键,并增长碳链。
羟醛缩合反应过程
1、碱与乙醛中的α-氢结合,形成一个烯醇负离子或负碳离子。
2、这个负离子作为亲核试剂,立即进攻另一个乙醛分子中的羰基碳原子,发生加成反应后生成一个中间负离子(烷氧负离子)。
3、烷氧负离子与水作用得到羟醛和OH。
稀酸也能使醛生成羟醛,但反应历程不同。
酸催化时,首先因质子的作用增强了碳氧双键的极化,使它变成烯醇式,随后发生加成反应得到羟醛。
生成物分子中的α-氢原子同时被羰基和β-碳上羟基所活化,因此只需稍微受热或酸的作用即发生分子内脱水而生成,α,β-不饱和醛。
凡是α-碳上有氢原子的β-羟基醛、酮都容易失去一分子水。
这是因为α-氢比较活泼,并且失水后的生成物具有共轭双键,因
此比较稳定。
除乙醛外,由其他醛所得到的羟醛缩合产物,都是在α-碳原子上带有支链的羟醛或烯醛。
羟醛缩合反应在有机合成上有重要的用途,它可以用来增长碳链,并能产生支链。
第3章羟醛缩合和有关的反应3.1 引言开链化合物的立体控制反应在现代有机化学中是一个备受关注的问题,已发展出许多有用的方法用于具有刚性构象的复杂分子(如大环内酯合多环醛抗生素)的立体控制合成。
醛醇缩合反应在生物合成中是一种基本的键形成反应,因而受到特别的注意。
醛醇反应,即亲核试剂与亲电的羰基基团(及类似基团)的缩合反应,是构建不对称C-C键的最简单的,同时能满足不对称有机合成方法学的最严格要求的一类化学转化。
在复杂分子的合成和在光学活性的小分子砌块的制备中,可以找到许多不对称醛醇缩合反应的实例[1].在复杂的天然产物的合成中,常常会遇到制备具有多个相邻的手性中心的中间体的任务。
这类化合物的最有效的合成策略,是那种在连接二个反应片断的同时又建立起毗邻立体中心的策略。
在每一个前述的策略中,希望对于相对的(syn/anti)以及绝对的(R/S)立体化学都能实行控制 . 已有许多研究者报道了非对映选择性(对映选择性)醛醇缩合反应的结果。
这些不对称醛醇缩合反应中的主要变化因素是金属抗衡离子、与这些离子键合的配体以及反应条件。
下述几种方法可用于对醛醇反应进行不对称控制:105(1)底物控制:非手性烯醇盐或烯丙基金属试剂对手性醛的加成(一般在α-位).在这种情况下,按照Cram-Felkin-Ahn规则由优势过渡态来决定非对映选择性[2].(2)试剂控制:手性烯醇盐或烯丙基金属试剂对非手性醛的加成.最通用的获得手性烯醇盐的方法是通过手性辅剂以酯、酰胺(噁唑啉)、酰亚胺(噁唑烷酮)或硼烯醇盐的形式结合;手性烯丙基金属试剂通常也与手性配体结合.(3)双不对称反应:手性烯醇盐或烯丙基金属试剂对手性醛的加成.当醛和试剂显示互补的面优先性(匹配对的情况)时,能够提高立体选择性;反之当它们的面优先性相反(错配对)时,立体选择性降低.当与合适的配体配位时,许多金属抗衡离于(诸如Li,Mg,Zr,B,AI,Sb,Si,Ti)在不对称醛醇缩合反应中能提供良好的立体选择性.锂或镁形成络台物,它们通过Cram-Felkin-Ahn规则或配位控制加成提供选择性.钛的应用得到了极巧妙的、多样性的好结果,它与手性配体络合提供对映选择性的转化.硼烯醇盐由于其高对映选择性的传递性质而被证明具有广泛的用途.杂双金属催化剂和双核中心催化剂既能活化亲核试剂又能活化亲电试剂,它们贯穿了本章的讨论内容.可以说,只是从20世纪80年代早期开始,本论题才获得显著的进展.在本章中,我们试图介绍金属烯醇盐和有关的烯丙基金属衍生物对碳基化合物的加成反应的一些最重要的进展,如图3.1中的途径A和途径B所示的。
106通常,醛与金属烯醇盐的醛醇缩合反应在产物分子中产生出二个新的手性中心,导致四个可能的立体异构体2a,2b,2c和2d(图3.2,图3.3)107以硼参与的醛醇缩合反应为例,羰基化合物从垂直角度接近烯醇盐化合物,反应经历六元环过渡态.二烷基硼的烯醇式具有相对来讲较短的硼-配体键及硼氧键,这是有利于过渡态中1,3竖键的相互作用,也有利于连接醛中的羰基的基团在过渡态中采取假平键状态.因此,从椅型环状过渡态3a—3d(Zimmerman-Traxler模型[3],见图3.3),我们可以得出结论,烯醇盐的几何结构可以演变为产物的2,3-立体化学:Z-烯醇倾向于产生syn-产物(Z→syn),而E-烯醇主要产生anti-产物(E→anti);产物中3-羟基的绝对构型可由烯醇盐接近羰基的方向决定:即Re→syn,Si→anti.有几个参数对于立体化学控制是极为关键的:(1)烯醇盐中取代基部分的空间大小;(2)选择合适的试剂; (3)烯醇化作用所选用的条件.因此,Masamune[4]设计了二种类型的醛醇缩合试剂,它们在醛醇缩合反应中导致相反的立体化学.在下列结构中,化合物4可用于获得anti-醛醇缩合产物,而化合物5则可用来合成syn-醛醇缩合产物(图3.4)。
当醛醇缩合试剂5在n-Bu2BOTf和Et3N存在下与醛反应时,可以高度非对映选择性地产生syn-醛醇缩合产物6(表3.1).1083.2 底物控制的醛醇缩合反应3.2.1噁唑烷酮作为手性辅剂参与的醛醇缩合型反应1964年,Mitsui[5]首次使用手性辅剂获得了不对称醛醇缩合反应立体控制的结果,虽然那时的立体选择性还不高(58%).在20世纪80年代早期出现了显著的改进,当时Evans[6]和Masamune[7]引入了一系列导致高立体选择性的手性辅剂.在与二烷基硼烯醇盐结合时,这些手性辅基诱导了高对映选择性的醛醇缩合反应.109由N-酰基噁唑烷酮(如7和8)(在第2章它们被称为Evans辅剂并被用于羰基的(α-烷基化反应)生成的手性硼烯醇盐由于其易于制备、优异的立体选择性、易于脱除和回收使用而倍受青睐[8].通常,可通过将N-酰基噁唑烷酮与二正丁基硼三氟甲磺酸盐和三乙胺在CH2Cl2中于-78℃一起处理以制备Z-硼烯醇盐.这样得到的烯醇盐在同样的温度下容易发生醛醇反应,产生syn-醛醇缩合产物,非对映选择性大于99%(图3.5).在此,硼抗衡离子对立体选择性起着重要的作用.在烯醇化步骤中用三乙胺代替二异丙基乙基胺得到的结果更好.但是将硼换成锂则导致立体选择性下降.有人假设,立体选择性来源于二齿金属(如硼)通过椅型过渡态9与噁唑烷酮的羰基和烯醇氧配位的结果(图3.5)[9].110如图3.5所说明的,Evans酰胺7和8能以相反的立体化学的方式与醛进行醛醇缩合反应.应指出的是,这点和在第2章(2.4.3节)所述的分子内配位所得到的产物在羰基α-位的立体化学相反。
将酰胺10和12用于反应时,可得到一对对映体11和13(当11 中R=Me时)(图3.6)[6].111双不对称诱导也可应用于醛醇缩合反应.当手性醛15用含有非手性硼参与的烯醇盐14处理时,以1.75:1的比例得到非对映体混合物;然而,让同样的醛15与由Evans辅剂8衍生的烯醇盐反应时,以极高的立体选择性获得syn-醛醇缩合产物16(属于匹配对,观察到600:1的非对映选择性).将16转化为Prelog-Djerassi内酯17就只是一种常规的反应了.即使在错配对的情况下,例如用另一个Evans辅剂7处理醛15(就立体选择性而言,与8比较,7起相反的功用),仍能以极满意的非对映选择性获得产物18(400:1).当然,从这两个反应得到的最终产物互为非对映异构体(图3.7).化合物17,是所谓的(+)-Prelog-Djerassi内酯酸,从酒霉素(Methmycin)或幂霉素(narbomycin)降解而得,具有一系列大环内酯抗生素共有的重要的结构特征,并成为发展许多新的立体选择性合成的焦点.化合物17的制备示于图3.8[10].从8(R=Me)出发,用醛处理硼烯醇盐,通过不对称醛醇缩合反应在C-2和C-2’以预期的立体化学合成了化合物20;用亲电试剂处理8的锂烯醇盐在C-5以预期的立体化学生成19.请注意醛醇缩合反应和α-烷基化反应的立体化学彼此正好相反.将19和20偶合得到最终产物17。
通过醛醇缩合反应,下列α-乙烯基-β-羟基亚酰胺21’也用于天然产物的全合成.在所有21参与的醛醇缩合反应的例子中,都能得到98%以上的d.e.值(图3.9)[11]。
1121133.2.2 吡咯烷作为手性辅剂如上述,在各种天然产物如大环内酯、离子载体抗生素和其它乙酰型化合物(acetogenics)中经常出现β-羟基羰基单元,这刺激了这些化合物的立体控制合成方法学的发展,其中最为成功的方法是使用醛醇缩合反应[12].带有反-2,5-二取代的吡咯烷部分(作为胺组份)的酰胺烯醇盐22,已证明是不对称烷基化反应[13]和酰化反应[14]的优良底物.与成功的烷基化和酰化反应结果不同,在醛醇缩合反应中使用其锂烯醇盐不能得到好的立体选择性(表3.2,项1).另一方面,从相应的Li烯醇盐和二氯化二(环戊二烯)合法制得的Zr烯醇盐[15]则表现出明显的好的立体选择性(表3.2, 2~5项).114研究表明,带有大基团配体的锆原子专一地位于Z-烯醇盐平面的底部半球,醛分子从同侧接近并与Zr原子配位,形成椅型过渡态,导致赤式醛醇的形成(图3.10,图3.11).对于Li烯醇盐,在烷基化反应或酰化反应中烷基卤或酰卤的进攻则直接在烯醇盐的顶面发生.具有立体要求的金属中心在调节醛醇115反应中的重要性是显而易见的.从所确定的缩合产物的立体化学可知,这个反应的不对称诱导和先前讨论中看到的烷基化反应或酰化反应的立体化学结果是相反的.在脯氨酸型手性辅剂中也遇到类似的情况.Evans发现,由脯氨醇酰胺衍生的锂烯醇盐在烷基化反应中显示优异的非对映面选择性(第2章,2.4.2节,脯氨酸型手性辅剂),但在醛醇缩合反应中使用脯氨酸酰胺的锂烯醇盐却不成功.有效果的手性试剂是锆烯醇盐,它可以从相应的锂烯醇盐通过与Cp2ZrCl2的金属交换而得到.例如,在锆烯醇盐参与的醛醇缩合反应中,能够获得极佳的不对称诱导效果,syn/anti选择性达96%~98%,非对映面选择性达50~200:1(图3.12).116117酰化产物25可通过烯醇盐24与酰氯反应制得。
有趣的是,用Zn(BH4)2和KBEt3H处理酰化的烯醇盐25可分别得到syn-或anti-26(图3.13,表3.3)[16]。
虽然起始步骤是α-酰化反应,但最终产物仍可被看作醛醇缩合产物。
3.2.3 氨基醇作为手性辅剂以上所介绍得醛醇缩合反应都是在碱性条件下进行的,其中烯醇盐作为反应活性中间体参与反应。
在碱性条件下的醛醇缩合反应,其副产物有双聚体,高聚体,以及α,β-不饱和羰基化合物等。
与公认的碳阴离子化学相反,Mukaiyama发展了另一种实用的方法,其中烯醇是关键的中间体。
他首次证明用TiCl4和硅烷基醇醚作为稳定的烯醇等价物也能进行酸催化的醛醇缩合反应[17]。
继而,他又发展了硼三氟甲烷磺酸盐参与的醛醇缩合反应,此反应通过形成甲酰烯醇醚而完成。
Mukaiyama首次报道,硅烷氧基烯烃作为必需的潜在烯醇盐等价物在Lewis酸活化剂存在下与醛反应,这个方法现在称为Mukaiyama醛醇缩合反应(图3.14)。
在Lewis酸存在下,通过醛与硅烷基烯酮缩醛(在动力学控制下从丙酸酯生成)反应,在多数情况下可得到anti-醛醇缩合产物[18]。
在各种Lewis酸中,TiCl4几乎是最佳选择[17b]。
118在手性硅烷基烯酮缩醛的“Mukaiyama醛醇缩合反应”中,另一种用于控制绝对立体化学的手性辅剂由N-甲基麻黄碱衍生而得[26]。
它成功地应用于α-甲基-β-羟基酯(91%~94% e. e.)[19],[26]、α-甲基-β-羟基醛(91% e. e.)[20]、α-肼基和α-氨基酸(78%~91% e.e.)[21]、α-甲基-δ-氧代酯(72%~75% e. e.)[19b]、顺和反-β-内酰胺(70%~96% e. e.)[22]、carbapenem抗生素[23]和其它天然产物[24]的对映选择性合成。