地中海贫血Thalassemia
- 格式:ppt
- 大小:4.78 MB
- 文档页数:51


关于地中海贫血的研究报告
地中海贫血(Thalassemia)是一种常见的遗传性血液病,主要影响红细胞的合成,导致患者体内缺乏正常的血红蛋白。
该病主要分为α地中海贫血和β地中海贫血两种类型,受到地中海地区高发的影响,因此得名。
以下是有关地中海贫血的研究报告的一些主要内容:
1. 疾病流行地区:地中海贫血主要流行于地中海盆地地区,包括地中海沿岸国家、中东地区和亚洲等地。
这些地区的高发率主要与该地区许多人存在与该疾病相关的遗传基因有关。
2. 发病原因:地中海贫血的发病主要与突变的遗传基因有关,其中α地中海贫血与α地中海贫血基因的缺陷相关,而β地中海贫血则与β地中海贫血基因的突变有关。
这些遗传基因突变导致血红蛋白在合成过程中发生障碍,进而导致患者生产出缺陷的血红蛋白。
3. 症状和诊断:地中海贫血患者常常表现出贫血、疲劳、黄疸等症状。
诊断地中海贫血主要依靠血液检查,包括完整血细胞计数、血红蛋白电泳分析等。
4. 治疗方法:地中海贫血目前尚无根治方法,主要通过输血和螯合治疗来维持患者的生命质量。
螯合治疗主要通过注射铁螯合剂来帮助患者排出过多的铁负荷,以减少铁负荷带来的伤害。
5. 遗传咨询和预防:由于地中海贫血是一种遗传性疾病,遗传
咨询在预防和管理方面起着重要作用。
遗传咨询可以帮助携带地中海贫血基因的夫妇了解风险,并提供相应的建议和指导。
总的来说,地中海贫血是一种遗传性的血液病,对患者的生活和健康产生了重要影响。
目前,研究主要关注于该病的发病机制、治疗方法和预防措施,但仍需要进一步的研究来提高对该疾病的认识和管理。

[键入文字]
地中海贫血能治吗地中海贫血的饮食
地中海贫血(Thalassemia)又称海洋性贫血。
是一组遗传性小细胞性溶血性贫血。
其共同特点是由于珠蛋白基因的缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少
或不能合成。
导致血红蛋白的组成成分改变,本组疾病的临床症状轻重不一,大多表
现为慢性进行性溶血性贫血。
下面小编为大家介绍地中海贫血能治吗及地中海贫血的
饮食。
地中海贫血(Thalassemia)又称海洋性贫血。
是一组遗传性小细胞性溶血性贫血。
其共同特点是由于珠蛋白基因的缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少或
不能合成。
导致血红蛋白的组成成分改变,本组疾病的临床症状轻重不一,大多表现
为慢性进行性溶血性贫血。
下面小编为大家介绍地中海贫血能治吗及地中海贫血的饮食。
地中海贫血能治吗
地中海贫血主要以预防为主,轻型无症状可不用治疗;重型地中海贫血要进行造血
干细胞移植,这种方法花费大,风险较高,但如果成功可以使重度贫患者摆脱对输血
的依赖及防止进一步铁沉着;若不进行造血干细胞移植,患者就只能依靠输血、长期
使用除铁剂维持生命,同时配合使用除铁剂,即便如此,长期输血,铁也会越来越多
地沉积在肝、脾等器官内,进而引起这些器官功能衰竭而导致患者死亡。
并且造血肝
细胞移植也存在相当大的危险性。
1。

地中海贫血的防治地中海贫血(Thalassemia)是一组由于珠蛋白基因的缺陷导致珠蛋白合成障碍所引起的遗传性溶血性疾病,以α和β地中海贫血最为常见。
主要见于地中海地区、东南亚地区和中国南方等地。
全世界7%的人群携带有地中海贫血基因,每年出生的重型地中海贫血患儿约50万,对家庭和社会带来很大的影响。
地中海贫血是通过人群干预即可以预防的遗传病之一。
通过公众教育,人群筛查,遗传咨询,对可能生育重型地中海贫血患儿的高危夫妇实施产前诊断和选择性流产,避免重型患儿出生是预防地中海贫血的重要措施。
一、地中海贫血的分子基础与临床表现:(一)β地中海贫血1. 分子缺陷:β珠蛋白基因簇位于11号染色体(11p15.5),包括ε,Gγ,Aγ,δ,β基因和2个假基因。
β珠蛋白基因的点突变,核苷酸缺失与插入,导致β珠蛋白链的合成减少或完全缺乏。
β0-地中海贫血:突变导致β 链的合成完全受抑制,无β链生成。
β+-地中海贫血:突变导致β链的合成部分受抑制,有少量β链生成。
非典型β地贫:分子缺陷位于β珠蛋白基因的启动区或位点控制区(LCR),导致β珠蛋白基因表达受影响。
β地中海贫血基因突变与临床类型:重型β地中海贫血(β-thalassemia major):纯合子:β0/β0,β+/β+双重杂合子:β0/β+中间型(β-thalassemia intermedia):纯合子:β+/β+双重杂合子:β0/β+合并α地中海贫血等轻型(β-thalassemia minor):杂合子β+, β0, δβ等。
2. 病理生理:当β链减少,多余的α链与γ链结合成为Hb F(α2γ2),使Hb F增高。
Hb F氧的亲和力高,使组织缺氧。
多余的α链沉积在幼红细胞和红细胞中形成包涵体,包涵体附着于红细胞膜使其变僵硬:幼红细胞在骨髓内被破坏,导致无效造血(ineffective erythropoiesis),而红细胞在通过外周血微循环时被破坏。

地中海贫血地中海贫血亦称为珠蛋白生成障碍性贫血或海洋性贫血(thalassemia),是一组遗传性溶血性贫血。
因珠蛋门基因缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少或不能合成,导致血红蛋白的组成成分发生改变,临床上主要表现为慢性溶血性贫血。
国外以地中海沿岸及东南亚各国多见,中国主要分布在长江以南各省,广东、广西、海南及四川等省(自治区)发病率较高,湖南、湖北及江两等省发病率稍低,北方各省少见。
胚胎期后组成珠蛋白的肽链有4种:α、β、γ、δ链,每种肽链各由相应的基因编码、。
因珠蛋白基缺失或点突变不同,肽链合成障碍各异,根据异常肽链类型将地中海贫血分为α、β、δβ、γ等类型,其中β和α地中海贫血较为常见。
β一地中海贫血:β一地中海贫血(简称β一地贫)是由于调控β珠蛋白的基因缺陷,导致β珠蛋白合成障碍的溶血性贫血。
1、发病遗传学人类β珠蛋白基因位于1 l号染色体短臂上(1 1p15.5),发生β一地贫的主要原因是基因的点突变,少数为基因缺失。
如果基因缺失或点突变导致β链的生成完全受到抑制,称为βo地贫;如果点突变仅使部分β链生成减少,则称为β+地贫。
β地贫基因变化复杂,已发现的点突变有200多种.1979年,中国发现第1个β一地贫基因点突变,迄今已累计发现相关基因点突变28种,最为常见的突变有6种:①β4l 42(‘TCTT),约占45%;②IVS一Ⅱ一654(c—T),约占24%;③1317(A—T),约占14%;④13—28 (A—T),约占9%;⑤[β7l一72(+A),约占2%;⑥[β26(G—A),约占2%。
根据酽或β+地贫基因的组合不同,临床表现为3型β一地贫:①重型:为βo基因的纯合子(βo/βo)、部分β+基因的纯合子(β+/β+)及部分βo和β+基因的双重杂合子(βo/β+);②中型:少数βo/βo,部分β+/β+,以及不典型β地贫杂合子、重型β地贫合并α或δβ地贫及某些变异型β地贫的纯合子等;③轻型:β+、βo、δβ地贫的杂合因子。

标准型地中海贫血一、引言地中海贫血(Thalassemia)是一种常见的遗传性血液疾病,主要在地中海沿岸和亚洲地区高发。
根据病情轻重,地中海贫血分为标准型、轻型、中间型和重型。
其中,标准型地中海贫血是一种较为常见的类型,对患者的生活质量和健康状况产生一定影响。
本文将详细介绍标准型地中海贫血的各个方面。
二、标准型地中海贫血的概述标准型地中海贫血,也称为β-地中海贫血,是一种由于血红蛋白β链缺失或减少而引起的贫血。
标准型地中海贫血通常在儿童期出现症状,表现为轻度至中度的贫血,无明显黄疸、肝脾肿大等症状。
患者通常需要定期输血以维持血红蛋白水平。
三、标准型地中海贫血的遗传学机制标准型地中海贫血的遗传学机制是由于血红蛋白β链基因的突变或缺失,导致血红蛋白β链合成障碍。
这些突变可以是小的缺失、点突变或大的基因片段缺失。
当一个个体从父母双方各继承一个异常β基因时,就会出现β链合成不平衡,导致红细胞易碎和寿命缩短,从而引发贫血。
四、标准型地中海贫血的诊断标准型地中海贫血的诊断主要依据临床表型和实验室检测结果。
常见的诊断方法包括:血红蛋白电泳、肽链分析、基因检测等。
通过这些检测方法,医生可以确定是否存在异常的血红蛋白成分或基因突变,进而确诊标准型地中海贫血。
五、标准型地中海贫血的治疗和管理标准型地中海贫血的治疗和管理主要包括以下方面:1.输血治疗:定期输血是维持患者血红蛋白水平的主要治疗方法。
根据患者的病情和年龄,医生会制定合适的输血计划。
然而,长期输血可能导致铁过载,需要进行去铁治疗。
2.去铁治疗:由于长期输血会导致铁过载,引发一系列并发症,因此去铁治疗至关重要。
常见的去铁治疗方法包括口服去铁剂和定期进行静脉去铁治疗。
3.饮食和生活方式调整:患者应注意保持均衡饮食,增加富含优质蛋白质、维生素和矿物质的食物摄入。
同时,适度运动和避免疲劳也有助于改善贫血症状。
4.预防感染:标准型地中海贫血患者免疫功能低下,容易感染。

常见地中海贫血的五种治疗方法地中海贫血(Thalassemia),又称地贫,是一种遗传性血液病。
它主要在地中海地区和周边国家发病率较高,因此得名。
地中海贫血的主要特点是患者体内的血红蛋白合成障碍,导致红细胞数量减少,造血功能受损,引发贫血。
地中海贫血需要长期的治疗,以下是几种常见的治疗方法。
1. 输血治疗:地中海贫血患者主要通过输血来治疗贫血。
输血可以补充患者体内的缺氧红细胞,提高血红蛋白含量,缓解贫血症状。
通常患者需要定期定量的输血治疗,旨在维持血红蛋白水平在一个较为正常的范围内。
然而,长期的输血治疗会导致患者体内铁离子积聚过多,因此需要联合使用螯合剂来帮助排除体内过多的铁离子。
2. 螯合剂治疗:由于输血会造成患者体内大量的铁负荷,所以地中海贫血患者需要使用螯合剂来帮助排除过多的铁离子。
螯合剂通过与体内的游离铁结合形成稳定的络合物,促使体内过量的铁离子通过尿液和粪便排出体外。
常用的螯合剂有海绵状铁剂(Desferrioxamine)和口服螯合剂(Deferiprone、Deferasirox)。
螯合剂的使用可以帮助地中海贫血患者减少体内的铁负荷,减轻后期对于器官损伤的风险。
3. 骨髓移植:对于适合骨髓移植的地中海贫血患者来说,这是一种常见的治疗方法。
骨髓移植可以完全根治地中海贫血,恢复正常的造血功能。
然而,由于骨髓移植具有风险性和成本高昂的特点,适合骨髓移植的患者比例相对较小。
骨髓移植需要一个合适的供者,并且在手术前后需要长期的免疫抑制剂药物治疗。
4. 基因治疗:随着基因工程技术的发展,基因治疗也成为治疗地中海贫血的潜在方法之一。
基因治疗通过将正常的基因序列导入患者的体细胞中,使其能够正常合成血红蛋白。
然而,基因治疗技术在地中海贫血的治疗上还处于实验阶段,并未广泛应用在临床实践中。
5. 心理治疗和支持性护理:地中海贫血患者需要面对长期的治疗和护理,因此需要心理治疗和支持性护理的帮助。
心理治疗可以帮助患者调整心态,提高生活质量。

海洋性贫血是什么:海洋性贫血又称地中海贫血(Thalassemia)。
是一组遗传性溶血性贫血。
其共同特点是由于珠蛋白基因的缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少或不能合成。
导致血红蛋白的组成成分改变,本组疾病的临床症状轻重不一,大多表现为慢性进行性溶血性贫血。
地中海贫血(在香港叫链状细胞性贫血)按照受累的氨基酸链来分类,主要有α地中海贫血(α链受累)和β地中海贫血(β链受累)。
也可按照一个或两个基因缺损来分为轻型或重型地中海贫血。
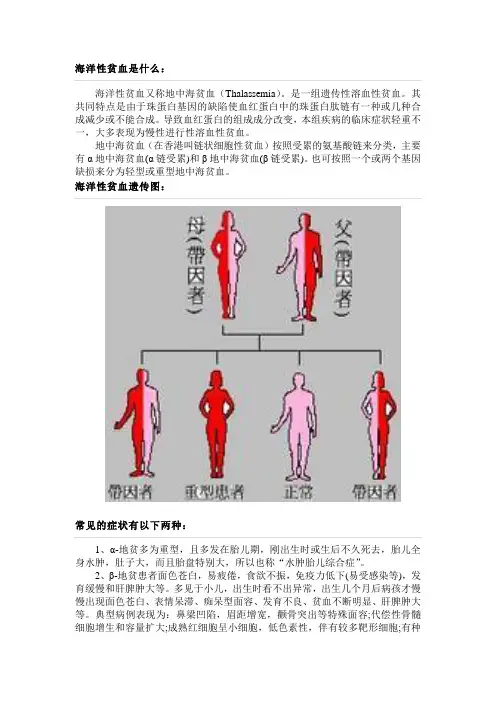
海洋性贫血遗传图:常见的症状有以下两种:1、α-地贫多为重型,且多发在胎儿期,刚出生时或生后不久死去,胎儿全身水肿,肚子大,而且胎盘特别大,所以也称“水肿胎儿综合症”。
2、β-地贫患者面色苍白,易疲倦,食欲不振,免疫力低下(易受感染等),发育缓慢和肝脾肿大等。
多见于小儿,出生时看不出异常,出生几个月后病孩才慢慢出现面色苍白、表情呆滞、痴呆型面容、发育不良、贫血不断明显、肝脾肿大等。
典型病例表现为:鼻梁凹陷,眉距增宽,颧骨突出等特殊面容;代偿性骨髓细胞增生和容量扩大;成熟红细胞呈小细胞,低色素性,伴有较多靶形细胞;有种族和家族史特征。
α地中海贫血多见于黑人(黑人中25%至少有一个基因缺陷),β地中海贫血多见于地中海地区或东南亚。
我国长江以南各省均有报道,以广东、广西、海南、四川、重庆等省区发病率较高,在北方较为少见。
若夫妻为同型地中海型贫血的带因者,每次怀孕,其子女有1/4的机会为正常,1/2的机会为带因者,另1/4的机会为重型地中海型贫血患者,因此,在遗传咨询及产前诊断方面,这是非常重要的疾病。
海洋性贫血的中医简介海洋性贫血在中医学中属“虚劳”、“黄疸”范畴。
自幼贫血,中焦受气,化血不足,更兼禀赋薄弱,阳不生阴,精血匮乏,水谷不能克消,精微反作水湿,阻遏胆液,浸渍肌肤为虚劳发黄之证,若气血阴阳不足,又见外邪客表,则可见虚实挟杂之征。
海洋性贫血三种类型(1)重型:出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型的表现是臀状头,长骨可骨折。

珠蛋白生成障碍性贫血病情说明指导书一、珠蛋白生成障碍性贫血概述珠蛋白生成障碍性贫血(thalassemia)原名地中海贫血、海洋性贫血,是由于一种或几种正常珠蛋白肽链合成障碍(部分或全部缺乏)而引起的遗传性溶血性疾病。
典型表现为皮肤苍白、乏力、易倦、黄疸、肝脾肿大、头颅大、鼻梁凹陷、眼距宽等,轻度患者多数预后较好,重度患者经积极治疗后症状会有所改善,但也有死亡的风险。
英文名称:thalassemia其它名称:地中海贫血、海洋性贫血相关中医疾病:暂无资料。
ICD 疾病编码:暂无编码。
疾病分类:血液系统疾病是否纳入医保:部分药物、耗材、诊治项目在医保报销范围,具体报销比例请咨询当地医院医保中心。
遗传性:有遗传性发病部位:全身常见症状:皮肤苍白、乏力、易倦、黄疸、肝脾肿大、头颅大、鼻梁凹陷、眼距宽主要病因:因一种或多种珠蛋白多肽链合成缺陷引起检查项目:体格检查、血常规、骨髓检查、血红蛋白电泳、铁代谢检查、X 线检查、基因诊断重要提醒:有本病家族史的女性,孕前应做遗传咨询评估,孕后应做产前检查和基因诊断,如有异常可在医生指导下终止妊娠。
临床分类:因涉及珠蛋白基因突变的种类及其影响繁多,故本病呈现高度异质性,根据受累的的珠蛋白链命名,分为α、β、γ、δ、δβ和εγδβ珠蛋白生成障碍性贫血,而α和β珠蛋白生成障碍性贫血最常见。
1、α珠蛋白生成障碍性贫血根据α基因缺失的数目和临床表现可分为静止型(1个α基因异常)、标准型(2个α基因异常)、血红蛋白 H 病(3个α基因异常)以及重型(4个α基因异常)。

轻度地中海贫血血常规指标轻度地中海贫血(Thalassemia minor)是一种遗传性血液病,主要特征是体内红细胞中球状部分的合成异常,导致血红蛋白合成缺陷。
轻度地中海贫血属于一种单基因疾病,患者通常表现为轻度贫血,但一般不需要特殊治疗。
1.血红蛋白(Hb):正常成年女性的血红蛋白浓度范围为120-160g/L,正常成年男性的血红蛋白浓度范围为130-180g/L。
轻度地中海贫血患者的血红蛋白浓度可以略低于正常范围,但通常仍处于正常的下限。
2.红细胞计数(RBC):正常成年女性的红细胞计数范围为3.8-5.1×10^12/L,正常成年男性的红细胞计数范围为4.4-5.8×10^12/L。
轻度地中海贫血患者的红细胞计数可以处于正常范围内或略低于正常范围。
3.红细胞平均体积(MCV):正常成年人的红细胞平均体积范围为80-100fL。
轻度地中海贫血患者的红细胞平均体积通常可以略低于正常范围。
4.红细胞分布宽度(RDW):红细胞分布宽度是反映红细胞大小不均匀程度的指标,正常范围为11.6-14.6%。
轻度地中海贫血患者通常会出现红细胞分布宽度的增加,即红细胞大小不均匀。
5.血小板计数(PLT):正常成年人的血小板计数范围为150-450×10^9/L。
轻度地中海贫血通常不会对血小板计数产生明显的影响。
6.白细胞计数(WBC):正常成人的白细胞计数范围为4-10×10^9/L。
轻度地中海贫血通常不会对白细胞计数产生明显的影响。
除了上述血常规指标外,还有一些其他指标可以用于评估轻度地中海贫血。
1. 血红蛋白电泳(Hb electrophoresis):血红蛋白电泳是一种常用的检查方法,可以用于鉴定不同类型地中海贫血。
轻度地中海贫血患者通常会显示血红蛋白A略偏低,而血红蛋白A2略增高。
2.铁代谢相关指标:轻度地中海贫血患者通常会表现为正常或轻度的铁代谢异常。
可以检测血清铁、转铁蛋白、铁饱和度和血清铁蛋白等指标来评估铁代谢情况。
【疾病名】小儿α-地中海贫血【英文名】pediatric alpha thalassaemia【缩写】【别名】小儿α-地贫;小儿α-海洋性贫血;小儿α-珠蛋白生成障碍贫血;小儿α-库利氏贫血;小儿α-库利贫血Mediterranean anemia;小儿α-Mediterranean disease【ICD号】D56.0【概述】地中海贫血(alpha thalassaemia)(简称地贫)又称海洋性贫血(thalassemia),据全国医学名词审定委员会规定应称为“珠蛋白生成障碍贫血”。
是由于一种或多种珠蛋白肽链合成受阻或完全抑制,导致Hb成分组成异常,引起慢性溶血性贫血。
根据不同类型的珠蛋白基因缺失或缺陷,而引起相应的珠蛋链合成受抑制情况不同,可将地贫分为α-地中海贫血;β-地中海贫血,δ-地中海贫血、γ-地中海贫血及少见的β-地中海贫血;以前2种类型常见。
各类地中海贫血之间又可互相组合,可与各种异常Hb组合(如HbE/β地中海贫血),这一组疾病又称地中海贫血综合征。
均属常染色体不完全显性遗传。
α-地中海贫血(α-mediterranean anemia)是由于α-珠蛋白基因的缺失或功能缺陷(点突变)而导致α-珠蛋白链合成障碍所引起的一组溶血性贫血。
【流行病学】从地中海沿岸的意大利、希腊、马耳他、塞浦路斯到东南亚各国均是本病多发区。
在我国的广东、广西、海南、云南、贵州、四川及香港等地区常见,发病率达10%~14%,黄河以南至长江流域,台湾、福建及西藏等省(区)均有病例报道。
患者以汉族为多,亦可见于回、傣、壮、苗和布依等少数民族。
【病因】属常染色体不完全显性遗传。
α-珠蛋白基因位于16号染色体短臂(16P13.33~p13.11~pter),总长约29kb,包含7个连锁的α类基因或假基因。
1.α-基因缺失 每条染色体上各有一对控制合成α-链的α-基因,因此每个细胞内有4个α-基因,可发生不同程度(1~4个)基因异常:(1)α-地贫(α地贫):若其中一条染色体上缺失一个α-基因组,则受控的α-链的合成部分受抑制,称为α-地贫(α地贫)。
α-地中海贫血缺失型基因
α-地中海贫血(α-thalassemia)是由于α珠蛋白基因缺失或突变导致的一种血红蛋白病。
缺失型α-地中海贫血基因是指α珠蛋白基因的部分或全部缺失,导致α珠蛋白链的合成减少或完全缺失。
α-地中海贫血的缺失型基因通常分为四种类型:α+-地中海贫血、α-地中海贫血、Hb-H病和中间型α-地中海贫血。
这些类型的区别在于缺失的基因数量和位置,以及它们对α珠蛋白链合成的影响程度。
α+-地中海贫血是由于一条染色体上的一个α基因缺失,导致α珠蛋白链的合成部分受抑制。
这种类型的地中海贫血通常是轻微的,可能没有明显的症状。
α-地中海贫血是由于一条染色体上的两个α基因都缺失,导致α珠蛋白链的合成完全受抑制。
这种类型的地中海贫血比α+-地中海贫血更为严重,可能导致贫血和其他症状。
Hb-H病(或中间型α-地中海贫血)是由于一对染色体上的四个α基因中缺失了三个,导致α珠蛋白链的合成大部分受抑制。
这种类型的地中海贫血症状介于α+-地中海贫血和α-地中海贫血之间。
缺失型α-地中海贫血基因可以通过一般治疗、应用药物、输血治疗和手术等方法进行治疗。
治疗方法的选择取决于病情的严重程度和患者的具体情况。
需要注意的是,地中海贫血是一种遗传性疾病,缺失型α-地中
海贫血基因可能会遗传给下一代。
因此,对于有地中海贫血家族史的人群,建议进行基因检测和遗传咨询,以便及早发现和治疗地中海贫血。
第1篇一、实验背景地中海贫血(Thalassemia)是一种由于珠蛋白基因缺陷引起的遗传性溶血性贫血病。
根据珠蛋白基因突变的不同,地中海贫血可分为α-地中海贫血和β-地中海贫血两大类。
本实验旨在通过分子生物学技术对疑似地中海贫血患者进行基因检测,以确定其基因型,为临床诊断和治疗提供依据。
二、实验目的1. 确定疑似地中海贫血患者的基因型。
2. 为临床诊断和治疗提供科学依据。
三、实验材料1. 样本:疑似地中海贫血患者的血液样本。
2. 试剂:DNA提取试剂盒、PCR试剂盒、DNA测序试剂盒等。
3. 仪器:PCR仪、电泳仪、测序仪等。
四、实验方法1. 样本DNA提取:采用DNA提取试剂盒提取疑似地中海贫血患者的血液样本DNA。
2. PCR扩增:根据α-地中海贫血和β-地中海贫血的基因突变位点,设计特异性引物,进行PCR扩增。
3. PCR产物检测:通过琼脂糖凝胶电泳检测PCR产物,观察条带情况。
4. DNA测序:对扩增产物进行DNA测序,确定基因突变位点。
5. 基因型分析:根据测序结果,分析疑似地中海贫血患者的基因型。
五、实验结果1. 样本DNA提取:成功提取疑似地中海贫血患者的血液样本DNA。
2. PCR扩增:成功扩增出α-地中海贫血和β-地中海贫血的基因片段。
3. PCR产物检测:电泳结果显示,扩增产物大小与预期相符。
4. DNA测序:测序结果显示,疑似地中海贫血患者的基因存在突变。
5. 基因型分析:根据测序结果,确定疑似地中海贫血患者的基因型为α-地中海贫血杂合子。
六、讨论与分析1. α-地中海贫血是一种常染色体隐性遗传病,主要由于α-珠蛋白基因缺陷引起。
本实验通过PCR和DNA测序技术,成功检测出疑似地中海贫血患者的基因突变,为临床诊断提供了依据。
2. α-地中海贫血的基因型可分为纯合子和杂合子。
本实验结果显示,疑似地中海贫血患者的基因型为α-地中海贫血杂合子,提示其可能表现为轻型α-地中海贫血或无症状携带者。
地中海贫血1.病名(包括英文、中文、OMIM编号)地中海贫血 thalassemiaOMIM编号: 141800(α-地中海贫血); 141900 (β-地中海贫血)2.疾病特点、发病率、诊断要点疾病特点:本病以血虚、黄疸和胁下积块为主要临床表现。
在婴儿期即发病者病情重,初期以肾精不足、肾精亏虚证为主,病久后影响其生长发育而见身材矮小、头大、前额门齿突出、鼻梁凹陷、两眼之间距离增大等特征性症状,并由于湿瘀之邪阻滞而见黄疸、肋下积块等症。
在成年后发病者逐渐出现血虚、黄疸及肋下积块等,一般病情较轻,遇有外感、用药不当、妊娠时病情可加重。
发病率:120-810/十万(α);40/十万(β)诊断要点:1)萎黄:姜黄是气血亏虚耗,失于荣养所致,表现为皮肤干黄无泽,伴头晕、心悸,与黄疸的根本区别在于白睛与小便均不黄。
2)黄汗:黄汗临床表现为汗出色黄染衣,但无黄疸之白睛色黄。
3.遗传方式与类型(包括各亚型)常染色体隐性 ARα-地中海贫血:HbBart胎儿水肿综合征,HbH,轻型和静止型β-地中海贫血:重型,中间型和轻型地中海贫血4.致病基因定位与克隆,基因功能等α-地中海贫血:HBA1/2,16p13.3,两组共4个拷贝,各有3个外显子,已报道点突变155种,大片段缺失突变43种,大片段插入突变1种,复杂重排突变4种,共计203种突变,以拷贝缺失突变为主。
β-地中海贫血:p15.5,3个外显子,已报道374种点突变,36种大片段缺失突变,1种大片段插入突变,8种复杂重排突变,共计419种突变,以基因点突变为主。
5.临床表现(病程、照片)a地中海贫血1.胎儿水肿综合征:胎盘大而易碎,胎儿全身水肿,轻度黄疸,皮肤可见出血点,心脏扩大;肺发育不全,胸腺缩小,肝脾肿大,腹水,胸腔及心包内积液,常于 30 ~ 40 周死于宫内, 其胎儿血中均为 HbBart 's ,而无 HbH 、 HbA2 及 HbF 。