拉曼光谱法指导原则
- 格式:pdf
- 大小:165.44 KB
- 文档页数:5

拉曼光谱的选律在外界电磁波的作用下,分子由某一个能级跃迁到另一个能级时,就产生了振动光谱,当两个振动态之间的跃迁几率为零时,这两个能级之间就不可能产生跃迁,也就不可能产生振动光谱,只有跃迁几率不为零的能级之间才可产生振动光谱。
跃迁几率问题是光谱理论的基本课题之一。
在外界电磁波的作用下,分子会产生跃迁行为,而分子出现在各种振动态上的几率不同。
分子所处的状态ψ只能是所有振动态的叠加状态。
因此,波函数ψ可写为下面线性组合的形式:ψ=ΣCKψK (1)故分子从ψn (未受外界作用时的状态)跃迁到ψm振动态的跃迁几率Pmn等于Pmn = |Cm|2=Cm* Cm (2)假定入射电磁波的波长比分子的线度大的多,利用含时微扰法,由经典电动力学电磁场的基本性质(拉格朗日方程)可计算出电偶极辐射的几率公式为:Pmn =4π2 I(ωMN)∣exmn∣2 /h2 (3)Pmn 称为电偶极辐射跃迁几率,原因在于忽略了入射电磁波在分子范围内的分布,或者说,在电偶极近似下认为电磁波在分子范围内是均匀分布的。
ex为感生电偶极矩。
所以拉曼光谱的机理本质是由在入射光电场作用下,在分子中所诱导的感生电偶极矩决定的。
由量子力学,矩阵元 xmn =∫ψm(o)* x ψn(0) dτ。
由(3)式,如果此积分为零,则相应状态之间的跃迁几率为零,我们称这两个状态是禁阻的;若此积分不为零,则它们之间的跃迁是容许的,称为容许跃迁。
那么,当ψm(o)和ψn(0)满足什么条件时,它们是容许或禁阻的跃迁呢?这些条件就称为振动光谱的选律。
若我们已知分子所属的点群,利用群的直积表示的方法,不用具体计算矩阵元 xmn,就可得到此积分为零或不为零的条件,即可确定拉曼光谱的普遍选律。
首先,我们给出一个结论:∫fAfB dτ这个积分中,只有被积函数在分子全部对称操作的作用下不变时,积分不为零,否则积分为零。
fAfB对于分子全部对称操作不变,即R1(fAfB)= fAfB,R2(fAfB)= fAfB,┄┄ Rn(fAfB)= fAfB 。
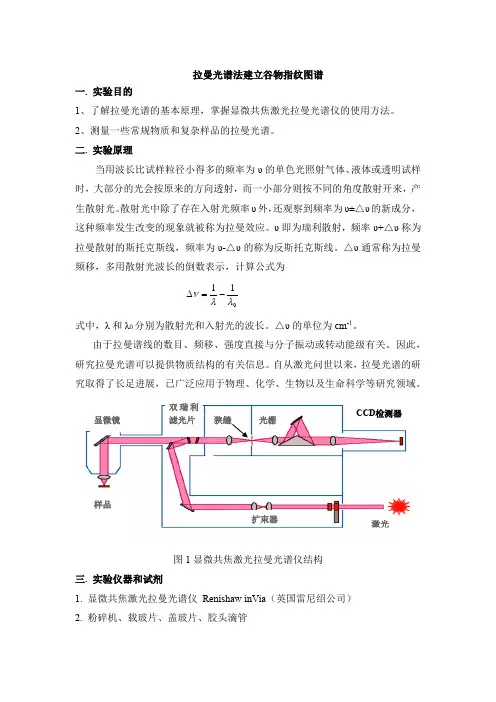
拉曼光谱法建立谷物指纹图谱一. 实验目的1、了解拉曼光谱的基本原理,掌握显微共焦激光拉曼光谱仪的使用方法。
2、测量一些常规物质和复杂样品的拉曼光谱。
二. 实验原理当用波长比试样粒径小得多的频率为υ的单色光照射气体、液体或透明试样时,大部分的光会按原来的方向透射,而一小部分则按不同的角度散射开来,产生散射光。
散射光中除了存在入射光频率υ外,还观察到频率为υ±△υ的新成分,这种频率发生改变的现象就被称为拉曼效应。
υ即为瑞利散射,频率υ+△υ称为拉曼散射的斯托克斯线,频率为υ-△υ的称为反斯托克斯线。
△υ通常称为拉曼频移,多用散射光波长的倒数表示,计算公式为011λλν-=∆式中,λ和λ0分别为散射光和入射光的波长。
△υ的单位为cm -1。
由于拉曼谱线的数目、频移、强度直接与分子振动或转动能级有关。
因此,研究拉曼光谱可以提供物质结构的有关信息。
自从激光问世以来,拉曼光谱的研究取得了长足进展,已广泛应用于物理、化学、生物以及生命科学等研究领域。
图1显微共焦激光拉曼光谱仪结构三. 实验仪器和试剂1. 显微共焦激光拉曼光谱仪 Renishaw inVia (英国雷尼绍公司)2. 粉碎机、载玻片、盖玻片、胶头滴管 显微镜 样品狭缝光栅扩束器3. 测试样品常规物质:CCl4,CH2Cl2复杂样品:不同淀粉类作物自备样品:不同材料的小挂件四. 实验步骤1. 打开主机和计算机电源,同时打开激光器后面的总电源开关,将仪器预热20分钟左右。
2. 自检.静态取谱(Static),中心520 Raman Shift cm-1, Advanced -> Pinhole 设为in。
使用硅片,用50 倍物镜,1 秒曝光时间,100%激光功率取谱。
使用曲线拟合(Curve fit)命令检查峰位,检验仪器状态。
3.样品拉曼光谱的测定将样品放置在载玻片上,盖上盖玻片,置于显微镜的载物台上,调节显微镜载物台的高度使得显微镜能够清晰地观察到样品表面(上2,下1)。

使用拉曼光谱仪进行材料分析的技巧拉曼光谱是一种非常有用的分析技术,可以用于研究和鉴定不同材料的化学成分以及结构信息。
本文将介绍使用拉曼光谱仪进行材料分析的一些技巧和注意事项。
一、拉曼光谱原理简介拉曼光谱是一种分析技术,利用激光照射样品时,光与样品分子之间发生相互作用,产生拉曼散射现象。
拉曼光谱可以提供物质分子的振动信息,从而确定其组成和结构。
拉曼光谱的特点是不需要对样品进行特殊处理,能够非破坏性地分析物质。
二、准备工作在使用拉曼光谱仪进行材料分析前,需要进行一些准备工作。
首先,确保光谱仪正常工作,激光器和检测器能够正常工作。
其次,准备好所需分析的样品,并确保样品表面干净,无尘或杂质。
此外,还需要检查实验室的环境条件,保持恒温和稳定的湿度,以减少外界因素对实验结果的影响。
三、样品的制备与处理对于固体样品,宜选择薄膜、颗粒或晶体,以获得较好的信号质量。
样品的表面应尽可能平坦、光洁,以确保激光能够均匀地照射样品表面。
对于液体样品,通常采用透明的玻璃容器进行分析,并确保容器内无气泡或杂质。
四、光谱测量参数选择在进行光谱测量之前,需要选择合适的测量参数。
首先是激光功率的选择,功率过高可能对样品造成热效应,功率过低可能导致信噪比低。
其次是积分时间的选择,根据实际情况确定积分时间,以充分获得信号质量。
此外,还需要确定光谱的测量范围,根据样品的特性和所需分析的信息进行选择。
五、数据处理与解读获得光谱数据后,需要进行数据处理与解读。
首先,对数据进行背景校正,以去除背景信号的干扰。
然后,进行光谱峰位的分析,确定峰位对应的振动模式。
此外,还可以进行峰位强度的定量分析,用于确定不同成分的含量和浓度。
最后,根据已有的参考谱与数据库进行对比,进行物质的鉴定和结构分析。
六、注意事项在使用拉曼光谱仪进行材料分析时,需要注意以下几点。
首先,避免样品受潮、受热或受光照射,以免影响实验结果。
其次,避免样品表面有杂质或污染物,以减少干扰。

光谱拉曼光谱定性检测原理光谱拉曼光谱是一种非常常用的光谱分析技术,可用于化学物质的定性和定量分析。
其原理基于拉曼散射现象,通过测量样品散射光谱中的拉曼散射光,可以获取关于样品的结构、成分和物理性质的信息。
本文将详细讨论光谱拉曼光谱定性检测的原理。
光谱拉曼光谱定性检测原理是基于分子与光相互作用而形成的。
当光与物质相互作用时,其中一部分光会被吸收,而另一部分光则会散射出去。
光谱拉曼光谱定性检测利用了被称为拉曼散射的特殊散射效应。
拉曼散射是指入射光与样品相互作用后,散射光谱中的部分光子被物质分子散射后的光子所吸收,同时也包含了新的光子能量和频率。
这些新产生的频率差异可以提供关于物质的结构、成分和物理性质的信息。
拉曼光谱分为两种类型:拉曼散射光的波长与入射光一致的称为弹性散射或瑞利散射,它只包含入射光的频率和振幅。
另一种是指拉曼散射光有不同频率或波长与入射光不同的称为非弹性散射,其中散射光的频率就是拉曼散射光的频率差异。
非弹性散射包括斯托克斯拉曼散射和反斯托克斯拉曼散射。
斯托克斯拉曼散射是指散射光频率低于入射光频率的情况,而反斯托克斯拉曼散射则是指散射光频率高于入射光频率的情况。
拉曼光谱的测量原理基于拉曼散射光的频率差异。
当光束照射到样品上时,一部分光被吸收,而其他部分光发生弱弱的拉曼散射。
散射的光通过光谱仪进行测量,得到散射光谱。
拉曼光谱通常使用激光作为入射光源,因为激光具有高强度和单色性,可以提供高质量的拉曼光谱信号。
拉曼光谱的分析过程中最重要的组成部分是拉曼散射光的检测和光谱仪分析。
检测系统一般包括一个光源、一个激光器、一个样品台和一个光探测器。
光谱仪分析是通过调整光线的色散来测量散射光的频率。
典型的光谱仪有拉曼光谱仪和光子散射光谱仪,其中拉曼光谱仪在分析振动频率时更常用,光子散射光谱仪则更多用于分析自旋激发频率。
拉曼光谱检测原理主要基于拉曼散射线的强度和频率与分子振动特性之间的关系。
分子振动模式涉及化学键的伸缩、弯曲、旋转等。

理论光谱学的拉曼光谱分析引言光谱学是研究物质与光的相互作用过程的学科。
其中,拉曼光谱分析是利用拉曼散射效应来研究物质的分子结构和化学成分的一种有效方法。
本文将从理论光谱学的角度出发,探究拉曼光谱分析的原理、仪器及应用。
1. 拉曼光谱分析的原理拉曼光谱是一种通过测量样品散射光的频移来获取样品分子的振动信息的技术。
其原理基于拉曼效应,即入射光与样品发生散射时,部分光子与样品分子相互作用后频率发生改变,从而产生拉曼散射光。
拉曼光谱分析的原理主要包括以下几点:1.1 可见光拉曼光谱可见光拉曼光谱是指样品在可见光范围内的拉曼光谱。
在可见光区域,拉曼散射光通常的能量与入射光相差很小,因此需要高灵敏的仪器进行检测。
1.2 红外拉曼光谱红外拉曼光谱是指样品在红外光范围内的拉曼光谱。
红外拉曼光谱可以用于表征样品的化学组成、结构和功能。
相比可见光拉曼光谱,红外拉曼光谱在分析材料的键合、分子构象和晶格振动等方面具有一定的优势。
1.3 拉曼光谱中的共振增强效应共振增强效应是指样品中某些特定振动模式的散射光谱强度远远大于其他振动模式的效应。
共振增强效应可以通过调整激发光的波长或变换样品的环境条件来实现。
2. 拉曼光谱仪的构成拉曼光谱仪是用于实施拉曼光谱分析的仪器装置。
它通常包括激光源、样品支承、散射光收集和检测、信号处理以及数据分析等模块。
2.1 激光源激光源是拉曼光谱仪的核心组件之一,它提供高亮度、高单色性的光束。
常用的激光源包括氩离子激光器、固体激光器、二极管激光器等。
2.2 样品支承样品支承模块是用于放置样品的部分。
样品可以采用液体、固体或气体形式。
常用的样品支承方式包括固体样品放在样品台上、液体样品放在带有透明窗口的样品池中。
2.3 散射光收集和检测散射光收集和检测模块主要用于采集样品的散射光,并将其转化为电信号。
常用的检测器包括光电二极管、光电倍增管等。
2.4 信号处理和数据分析信号处理和数据分析模块用于处理和分析采集到的散射光信号。


拉曼光谱实验注意事项一、样品准备在进行拉曼光谱实验前,需要充分了解样品的性质和特点,以便选择合适的实验条件和样品处理方式。
以下是一些需要注意的事项:1. 样品应具有代表性,能够反映所研究对象的特征或性质。
2. 对于不透明的样品,需要将其表面打磨或抛光,以便激光能够穿透样品并获得清晰的拉曼光谱。
3. 对于液体样品,需要将其稀释至一定浓度,以便在拉曼光谱中获得清晰的信号。
4. 对于气体样品,需要确保样品纯净且无杂质,以免干扰拉曼光谱的测量结果。
5. 对于固体样品,需要将其固定在样品台上,以确保其稳定性和可靠性。
二、实验环境拉曼光谱实验需要在一定的实验环境下进行,以确保实验结果的准确性和可靠性。
以下是一些需要注意的事项:1. 实验室应保持干燥、清洁、无尘,避免样品受潮、污染或损坏。
2. 实验室的温度和湿度应保持恒定,以确保仪器的稳定性和可靠性。
3. 实验室内应避免强光直接照射仪器和样品,以免干扰实验结果。
4. 实验室内应保持安静,避免噪音干扰实验结果。
5. 实验室内的电源和接地应符合仪器要求,以确保仪器的正常运行和安全。
三、激光安全拉曼光谱实验中使用的激光具有较高的能量和亮度,需要注意激光安全问题。
以下是一些需要注意的事项:1. 实验时应佩戴合适的防护眼镜或防护面罩,以避免激光直接照射到眼睛或面部。
2. 实验时应避免激光照射到皮肤或衣物上,以免造成损伤或烧伤。
3. 实验时应保持仪器整洁、干净,避免激光照射到灰尘或其他污染物上,以免造成火灾或爆炸等安全事故。
4. 实验时应严格按照仪器操作规程进行操作,避免误操作导致激光能量过高或过低等不安全因素。
5. 实验时应定期检查仪器的安全性能和防护措施,确保其正常、可靠地运行。
四、仪器校准拉曼光谱实验中使用的仪器需要进行定期校准和维护,以确保实验结果的准确性和可靠性。
以下是一些需要注意的事项:1. 仪器应定期进行校准和维护,以确保其正常、可靠地运行。
2. 在进行仪器校准和维护前,需要充分了解仪器的原理、结构、性能和使用方法等方面的知识。

国外用拉曼光谱对药品进行鉴别的方法拉曼光谱是一种快速而准确的分析技术,被广泛应用于药品鉴别、质量控制和验证等领域。
下面是国外使用拉曼光谱对药品进行鉴别的方法和步骤:
1. 样品准备
样品应该是粉末或晶体状的,必须无水或干燥,在样品之间保留足够的距离,以避免相互污染或交叉感染,同时应该确保透过玻璃或石英盘时没有过多的背景信号。
2. 拉曼光谱仪设置
使用适当的激光波长,一般是532或785纳米。
选择合适的光谱仪,例如RamanStation 400 或 QE Pro-Raman Spectrometer。
将样品放在透明的玻璃或石英盘上,并确保样品位置准确。
3. 数据采集
使用合适的软件,在样品表面扫描激光,并记录回散发射光谱图。
确保扫描速度适中,以确保数据质量和准确性。
将每个样品的光谱记录下来进行比较。
4. 数据解释
使用化学软件对光谱图进行分析和解释,并与数据库中的标准谱图进行比对,以确定药品的成分和质量是否符合要求。
如果药物检测出错或质量不符要求,则会重新进行分析或进行其他检测方法。
总之,拉曼光谱对药品鉴别是一种高效、快速、准确的方法,它可以准确识别有毒或低质药品,并帮助鉴定质量问题所在,从而有效地保护了公众的健康和安全。

使用拉曼光谱技术的注意事项近年来,拉曼光谱技术在科学研究、工业分析和材料检测等领域得到越来越广泛的应用。
作为一种非侵入性的分析方法,拉曼光谱技术可以提供物质的分子结构、化学组成和晶体形态等信息。
然而,要想获得准确可靠的拉曼光谱测试结果,我们需要在实验过程中注意一些事项,以确保实验的稳定性和可重复性。
首先,选择正确的激发源和探测器是至关重要的。
激发源的选择应根据待测物质的性质和要求而定。
常用的激发源包括激光器、氙灯和LED等。
激发源的功率和波长选择应使样品能够产生足够的拉曼散射信号,并避免光敏样品的损坏。
同样,探测器的选择也应根据拉曼光谱信号的强弱和要求进行调整,以保证信号的清晰度和准确性。
其次,样品的制备对于拉曼光谱测试的结果至关重要。
样品表面的平整度以及杂质的存在都会对拉曼光谱信号的质量产生影响。
因此,在样品制备过程中,应特别注意避免表面粗糙度、振动和尘埃等因素的干扰。
此外,对于液态样品,还应注意溶液的浓度和pH值等因素,以确保拉曼光谱测试的准确性。
第三,温度和湿度的控制也是使用拉曼光谱技术时需要重视的因素。
温度对于样品的相变和分子振动状态都有着重要影响。
因此,为了得到准确的拉曼光谱,我们需要在实验室中控制室温,并尽量减小温度的波动。
类似地,湿度对于一些特定的样品也会有一定的影响,可能引起湿度变化引起信号干扰和噪音。
因此,在拉曼光谱测试过程中,应尽量保持适宜的湿度水平,以确保实验结果的稳定性。
最后,拉曼光谱测量的数据处理也是使用该技术时需要特别关注的方面。
在进行拉曼光谱测量时,我们得到的是一个光谱图像,其中包含了大量的峰和谷。
要将这些数据转化为有关样品的有用信息,需要进行数据处理和谱图解释。
这包括标定光谱、消除背景噪音、峰拟合、光譜重建和数据解释等步骤。
通过正确的数据处理和分析,我们可以得到更加准确和可靠的拉曼光谱结果。
综上所述,使用拉曼光谱技术需要注意激发源和探测器的选择,严格控制样品的制备过程,关注温湿度的调控,以及合理处理和分析实验数据。

使用拉曼光谱仪时的注意事项光谱仪操作规程拉曼光谱是一种散射光谱,拉曼光谱分析法是基于拉曼散射效应,对与入射光频率不同的散射光谱进行分析以得到分子振动、转动方面信息,并应用于分子结构讨论的一种分析方法。
由分子振动、固体中光学声子等激发与激光相互作用产生的非弹性散射称为拉曼散射。
1、不要在潮湿或者高温的环境下操作仪器,避开各类磁场的干扰,这样才能检测出更高的精度。
2、测试样品前确定要请仪器厂商的技术人员现场演示,并且对技术人员做好操作前的培训。
仪器检测会要做放样检测,所以还需要做好防辐射。
3、假如长期不使用拉曼光谱仪时,请将主电源开关关掉,将头座拔掉,并将室外机用包装物包好防止灰尘进入。
并用中性清洁剂清洗空调过滤器。
4、使用中严格依照操作规程进行操作,在碰到故障时应首先咨询售后,仪器在送回维护和修理中心前客户应认真检查机器是否有物理性损坏。
5、拉曼光谱分析仪常用的试样制备方法是溴化钾(KBr)压片法,因此为削减对测定的影响,所用KBr应为光学试剂级,至少也要分析纯级。
6、如所用的是单光朿型傅里叶红外分光光度计,试验室里CO2含量不能太高,因此试验室里的人数应尽量少,无关人员不要进入,还要注意适当通风换气。
7、为防止仪器受潮而影响使用寿命,红外试验室应常常保持干燥。
即使仪器不用,也应当每周开机至少两次,定期除湿。
光纤光谱仪原理光路结构外部触发功能光纤光谱仪原理的优点在于系统的模块化和快捷性。
美国海洋光学公司的微小型光纤光谱仪的测量速度特别快,使得它可以用于在线分析。
而且由于它选用低成本的通用探测器,所以光谱仪的成本也大大降低,从而大大扩展了它的应用领域。
系列光纤光谱仪接受微型光机平台,尺寸只出名片大小,不仅便利携带,更能轻易地集成到各类光谱分析设备中。
光纤光谱仪原理优化的光路结构:接受交叉非对称C—T光路结构,中心波长除去彗差;内表平面抑制杂散光处理,除去了二级衍射的影响。
光纤光谱仪原理外部触发功能:光谱采样包括四种触发方式:正常模式,软件触发模式,同步触发模式和外部硬件触发模式。

第5章拉曼光谱分析法拉曼光谱分析法是一种基于拉曼散射原理的光谱分析技术。
该技术利用物质分子产生的拉曼散射光谱,通过测定光谱的频移来分析样品的成分和结构信息。
相比于传统的红外光谱分析法,拉曼光谱分析法具有高分辨率、非破坏性等优点,因此在各个领域得到了广泛应用。
拉曼光谱的基本原理是:当物质受到入射光的作用后,部分光子的能量被物质分子吸收,并在分子的振动和转动过程中增加或减少了能量,此时吸收光谱已经发生了位移。
通过分析这种能量的位移,可以获取样品的结构和成分信息。
通过拉曼光谱分析法,可以对各种物质进行非破坏性的分析。
例如,在化学领域,可以利用拉曼光谱分析法来确定化学反应中的中间产物和催化剂,以及分析有机化合物的结构。
在生物领域,可以用来研究生物分子之间的相互作用和结构变化。
在材料科学领域,可以分析材料的晶格结构和缺陷情况。
在环境领域,可以用来分析水和空气中的污染物。
拉曼光谱分析法的实施一般需要一个拉曼光谱仪。
这种仪器由激光系统、照射样品的光学系统、通过光学系统收集和分析拉曼散射光的系统以及数据处理系统组成。
首先,激光器产生一个单色激光束,照射到样品上。
样品散射的光经过光学系统聚焦到检测器上,并通过光电倍增管转化为电信号。
最后,数据处理系统会对电信号进行处理,得到拉曼光谱图。
在拉曼光谱分析法中,有两种常用的技术:常规拉曼光谱和表面增强拉曼光谱(SERS)。
常规拉曼光谱的灵敏度较低,需要较高的浓度才能获得良好的信噪比。
而SERS可以通过将样品与金属表面接触来放大拉曼信号,因此可以在极低浓度下进行分析。
总之,拉曼光谱分析法是一种高分辨率且非破坏性的光谱分析技术。
它在不同领域中有着广泛的应用,能够为我们提供样品的结构和成分信息。
随着技术的不断进步,相信拉曼光谱分析法将会在更多的领域得到应用。
使用拉曼光谱仪的技巧拉曼光谱仪是一种广泛应用于材料科学、生物化学和环境分析等领域的仪器。
通过测量样品中光散射光的频移,拉曼光谱仪可以提供有关样品的结构和组成的信息。
然而,为了获得准确可靠的结果,使用拉曼光谱仪时需要一定的技巧和注意事项。
首先,正确选择激发光源是使用拉曼光谱仪的关键。
激发光源的选择应基于样品的性质和所需的测量目的。
常用的激发光源包括激光二极管和氩离子激光器。
激光二极管适用于对样品表面进行非侵入性测量,而氩离子激光器可提供更高的能量和更广泛的光谱范围。
在选择激发光源时,还需要考虑光强、波长和稳定性等因素。
其次,样品的制备和处理对拉曼光谱的测量结果也有重要影响。
在制备样品时,应确保样品的纯度和均匀性,并避免污染和杂质的引入。
对于固体样品,可以通过将样品粉碎、压片或制备薄膜等方式进行处理。
对于液体样品,可以通过过滤、稀释或调整pH值等方法进行处理。
在处理样品时,还需要注意避免光源和样品之间的相互作用,以及干扰因素对拉曼光谱的影响。
第三,仪器的校准和调试是确保拉曼光谱仪正常工作的关键步骤。
在使用拉曼光谱仪之前,必须首先进行仪器的校准。
校准包括对激发光源的能量进行调整,校准波长范围和光强等参数。
在校准过程中,可以使用标准物质进行比对和修正。
此外,还需要定期检查仪器的光学系统,确保其正常运行和准确度。
如果发现任何问题,应及时进行维修和调试,以确保拉曼光谱仪的精确性和可靠性。
最后,数据的处理和分析是使用拉曼光谱仪的最后一步。
在进行数据处理时,可以使用专业的数据处理软件,如Origin、Matlab等。
这些软件可以对拉曼光谱的峰位、强度和形状进行拟合和分析,以提取样品的结构和组成信息。
此外,还可以进行数据展示和比较,达到更全面和深入的分析目的。
在使用拉曼光谱仪时,还需要注意一些常见的技巧和注意事项。
首先,应避免使用过高的激发光强度,以防止样品的热效应和光漂白现象。
其次,应注意保持样品和仪器的清洁和干燥,以减少干扰和噪声的影响。
拉曼光谱对样品的要求
拉曼光谱是一种分析化学技术,通过测量样品散射的拉曼散射光谱来获得关于样品的化学成分和结构信息。
为了获得高质量的拉曼光谱数据,样品需要满足一些要求和条件,以下是一些主要的要求:
1.清洁表面:样品表面应该是干净的,没有杂质、油
脂或污渍,因为这些可以干扰拉曼信号。
2.透明性:拉曼光谱对样品的透明性要求较高。
对于
不透明样品,需要使用透明基底或透明容器来容纳
样品,并测量样品与基底或容器之间的界面。
3.非荧光性:荧光信号会干扰拉曼信号,因此最好选
择不具有荧光性的样品。
如果样品本身具有荧光
性,可以考虑使用抑制荧光的方法,如使用激发光
源的波长远离激发荧光的波长。
4.均匀性:样品应该是均匀的,以确保获得代表性的
拉曼信号。
不均匀性可能导致信号的变异。
5.适当的浓度:样品的浓度应该在合适的范围内,通
常在微摩尔(μM)到毫摩尔(mM)之间。
过低的浓
度可能导致信噪比较低,而过高的浓度可能引起光
吸收或光散射,干扰拉曼信号。
6.样品的物理状态:样品可以是气体、液体或固体,
但需要适当的实验条件来适应样品的物理状态。
例
如,对于液体样品,可以使用适当的透明容器,并
确保样品没有结块或沉淀。
7.选择适当的激发光源:根据样品的特性选择合适的
激发光源波长。
不同的样品可能对不同波长的光散
射更敏感。
总之,拉曼光谱要求样品具有透明性、非荧光性、均匀性和适当的浓度,以获得准确的分析结果。
在进行拉曼光谱分析之前,通常需要对样品进行适当的准备和优化实验条件,以满足这些要求。
拉曼光谱制样要求嘿,朋友们!今天咱来聊聊拉曼光谱制样那些事儿。
这可真是个有意思的活儿呀!你想想看,拉曼光谱就像是一个神奇的眼睛,能看穿物质的本质呢。
但要让它好好工作,制样可得讲究咯!就好像你要给客人准备一顿丰盛的饭菜,食材得选好、处理得当吧。
先说固体样品。
哎呀呀,这就像是雕琢一块美玉,得小心翼翼的。
把它打磨得平平整整、干干净净的,可不能有啥杂质呀,不然拉曼光谱这个“眼睛”可要被遮住啦!你得把它切成合适的大小,太大了不行,太小了也不好找呀。
就像你穿衣服,得合身才好看嘛!然后是液体样品呢。
这就像是调一杯美酒,得把握好浓度和纯净度。
不能太稀了,也不能太稠了,得恰到好处。
要是里面有啥小颗粒啥的,那不就成了“浑酒”啦,拉曼光谱可就看不清啦!所以过滤这一步可千万不能马虎呀。
还有啊,制样的时候环境也很重要哦!不能在一个脏兮兮、乱糟糟的地方弄吧。
那可不行,就好比你在一个满是灰尘的房间里做饭,做出来的能好吃吗?得找个干净、整洁的地方,让样品也感觉舒舒服服的。
再说说样品的保存。
这就像把宝贝藏好一样,不能让它受到一点伤害。
要放在合适的容器里,密封好,别让它受潮啦、被污染啦。
不然等你要用的时候,发现坏了,那不就傻眼啦!你说制样麻烦不麻烦?当然麻烦啦!但这就是科学呀,没有这些细致的工作,哪能得出准确的结果呢?就像建房子,没有牢固的地基,哪能有漂亮的高楼大厦呢?总之啊,拉曼光谱制样可不是随随便便就能搞定的事儿。
得用心、细心、耐心,就像对待自己最珍贵的宝贝一样。
只有这样,才能让拉曼光谱发挥出它最大的威力,帮我们解开物质世界的奥秘呀!所以朋友们,可别小瞧了这制样的工作哦,它可是至关重要的一步呢!。
拉曼光谱制样注意点拉曼光谱是一种非破坏性的分析方法,其应用范围广泛,可以用于化学、物理、生命科学等领域的表面或者内部成分分析。
然而在进行拉曼光谱实验时,样品的制备过程是至关重要的,粗糙的制备方法会导致实验数据的失真或者不可靠性。
因此,在进行拉曼光谱分析前,需要认真把握拉曼光谱制样的注意点。
以下是几个制样中需要重点注意的方面:1. 样品的来源和性质样品来源和性质直接影响制备的方式和步骤。
不同的样品可能需要不同的制样方式,例如不同的样品制备过程可能需要考虑其中的结构、晶型、杂质含量等等。
因此,在制备过程中,需要准确了解样品的来源和性质,并严格按照相关要求进行制备。
2. 样品的制备过程样品制备需要注意的步骤包括样品的清洁、样品的粉碎和平均化、样品涂覆等。
清洁样品避免了杂质的干扰,而样品的粉碎和平均化可以使样品更加均匀,提高数据的可靠性。
涂覆时要避免产生气泡和不同厚度的涂层,否则会对实验的结果造成不良影响。
3. 样品的储存和保养制备好的样品需要适当的储存和保养,以保证其稳定性和一致性。
在储存过程中,可以考虑将样品放置在干燥、防潮、避光的环境中,保持样品内部结构的稳定性。
并且,制备好的样品需要配备相应的标签或者记录,以便进行准确的实验记录和数据分析。
4. 样品制备时在场环境的保持在进行拉曼光谱实验的制样过程中,注意保持制样现场的实验环境的整洁和稳定。
适当的调节实验室温度和湿度可以保持一定的制样质量,以避免实验结果的不稳定性和偏差。
同时,制样现场应保持干燥,避免有杂质进入样品中,影响实验数据的真实性和准确性。
综上所述,拉曼光谱分析是一种高效、准确的表面或者内部成分分析方法,然而在进行拉曼光谱分析前,需要前期的制样准备工作。
样品的来源和性质、制备过程、储存和保养、在场环境的保持是四个重要的制样注意点。
当我们在制备过程中严格按照要求进行制备和检验时,才能得到准确、可靠的拉曼光谱数据,提供有力的分析依据。