结构化学之计算化学
- 格式:doc
- 大小:32.50 KB
- 文档页数:3


计算题 ( 附答案 )1. 5 分用透射电子显微镜摄取某化合物的选区电子衍射图,加速电压为200?kV ,计算电子加速后运动时的波长。
2. 10 分限制在一个平面中运动的两个质量分别为m 1和m 2的质点 , 用长为R 的、没有质量的棒连接着, 构成一个刚性转子。
(1) 建立此转子的Schrödinger 方程, 并求能量的本征值和归一化的本征函数;(2) 求该转子基态的角动量平均值。
已知角动量算符 M ˆ=M ˆz =-i π2h φ∂∂。
3. 10 分分子CH 2CHCHCHCHCHCHCH 2中的π电子可视为在长为8R c-c 的一维势箱中运动的自由粒子。
分子的最低激发能是多少?它从白色光中吸收什么颜色的光;它在白光中显示什么颜色? (已知 R c-c=140 pm)4. 10 分试证明三维势箱中粒子的平均位置为(a /2, b /2, c /2)。
5. 10 分①丁二烯 和②维生素A 分别为无色和橘黄色,如何用自由电子模型定性解释。
②已知丁二烯碳碳键长为1.35×10-10?nm(平均值),维生素A 中共轭体系的总长度为1.05?nm(实验值)。
6. 10 分已知 Li 2+ 的 1s 波函数为32130s 1e 27a r -α⎥⎦⎤⎢⎣⎡π=ψ(1)计算 1s 电子径向分布函数最大值离核的距离;(2)计算 1s 电子离核平均距离;(3)计算 1s 电子概率密度最大处离核的距离。
(10!d e +∞-=⎰n ax n a n x x )7. 10 分已知类氢离子 sp 3杂化轨道的一个波函数为:x p s 3sp 2321φφψ+= 求这个状态的角动量平均值的大小。
8. 10 分电离1mol 自由铜原子得1mol Cu +,需能量为746.2 kJ ,而由铜晶体电离获1 mol Cu +仅消耗 434.1 kJ 能量。
(1) 说明上述两电离过程所需能量不同的原因;(2) 电离 1 mol 铜晶体所需照射光的最大波长是多少?(3) 升高温度能否大大改变上述两电离过程所需能量之差?9. 5 分波函数具有节面正是微粒运动的波动性的表现。

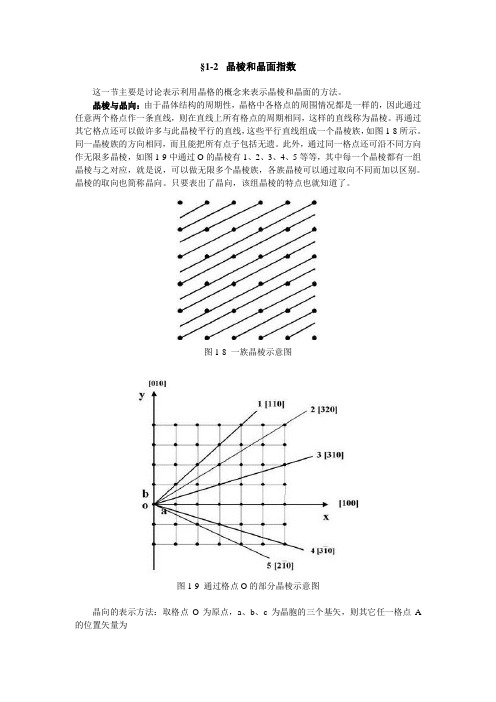
§1-2 晶棱和晶面指数这一节主要是讨论表示利用晶格的概念来表示晶棱和晶面的方法。
晶棱与晶向:由于晶体结构的周期性,晶格中各格点的周围情况都是一样的,因此通过任意两个格点作一条直线,则在直线上所有格点的周期相同,这样的直线称为晶棱。
再通过其它格点还可以做许多与此晶棱平行的直线,这些平行直线组成一个晶棱族,如图1-8所示。
同一晶棱族的方向相同,而且能把所有点子包括无遗。
此外,通过同一格点还可沿不同方向作无限多晶棱,如图1-9中通过O的晶棱有1、2、3、4、5等等,其中每一个晶棱都有一组晶棱与之对应,就是说,可以做无限多个晶棱族,各族晶棱可以通过取向不同而加以区别。
晶棱的取向也简称晶向。
只要表出了晶向,该组晶棱的特点也就知道了。
图1-8 一族晶棱示意图图1-9 通过格点O的部分晶棱示意图晶向的表示方法:取格点O为原点,a、b、c为晶胞的三个基矢,则其它任一格点A 的位置矢量为式中l1、l2、l3为整数(或有理数)。
取l1、l2、l3的互质比,即l1:l2:l3来表示晶棱OA 的方向,通常不直接用比例记号,该用方括号[l1l2l3]表示。
例如在图1-9中,晶棱1上A点为l1=1,l2=1,l3=0;B点为l1=2,l2=2,l3=0;比值为:l1:l2:l3=1:1:0=2:2:0,由此可得晶棱1的方向为[110]。
同理可得晶棱2的方向为[320],晶棱4的方向为[30],其中记号“”代表“-1”。
三个晶轴a、b、c的方向分别为[100]、[010]、[001](c轴与图平面垂直,未画出)。
晶面与晶面指数:晶格中,还可以从各个方向上划分成无限多平面,即晶面族,如图1-10所示。
同一族晶面中,彼此距离相等,方向相同,格点在晶面上的分布也相同。
晶体的表面也是晶面,通常应该是原子面密度比较大的面。
现在问题是如何表示这些晶面族的方向。
图1-10 部分晶面族示意图从立体几何中知道,要描述一个平面的方向,就是表示出这个平面在三个坐标轴上的截距。

第三章 双原子分子结构3.1 +2H 的结构及共价键的本质基本内容—、定核近似和+2H 的薛定谔方程A BRe r e r e m H b a 02020*******ˆπεπεπε+--∇-= 我们常采用原子单位:单位长度:Pm e m h a e 9177.524422200==ππε(玻尔半径)单位质量:me=9.1095×10-31Kg (电子质量) 单位电荷:e=1.60219×10-19C (电子电量) 单位能量:024a e πε=27.2116eV单位角动量: =1.0546×10-34 J.S 单位介电常数:04πε=1采用原子单位、+2H 的哈密顿算符为:Rr r Hba 11121ˆ2+--∇-=其薛定谔方程为:ψψE Rr r b a =+--∇-)11121(2,式中E 、ψ分别为+2H 的波函数和能量。
二、变分原理及性线变分法 1. 变分原理对于任意一个品优波函数ψ,用体系的Hˆ算符求得的能量平均值将大于或接近等于体系基态的能量E 0即:*ˆ*E d d H E ≥>=<⎰⎰τψψτψψ 据此原理,利用求极值方法调节参数,找出能量最低时对应的波函数,即为和体系基态相近似的波函数。
2. 线性变分法在量化计算中,广泛采用的是线性变分函数,它是满足体系边界条件的 个线性无关的函数m φφφ,,,21 的线性组合:m m C C C φφφψ+++= 2211采用线性变分函数的变分法叫线性变分法。
根据变分原理求得使E 最低的一组组合系数Ci⎰⎰++++++++++++=τφφφφφφτφφφφφφd C C C C C C d C C C H C C C E m m mm m m m m ))(()(ˆ)(2211***2*2*1*12211***2*2*1*1mC EC E C E ∂∂==∂∂=∂∂ 21=0 由此得一组求解Ci 的m 个联立方程称为久期方程,运用线性代数法求得m 套非零解,由其中与最低E 相对应的一套解C 1,C 2,……,C m 便可组成基态分子轨道波函数,所对应的E 便是基态能量近似值。

例:分别计算丁二烯处于基态和第一激发态时的电子密 度、键级、自由价,画出分子图。
分析: 丁二烯基态电子排布: 丁二烯第一激发态电子排布:基态: (1) 电荷密度:C1:q1=2×0.37172+2×0.60152=1.0 C2:q2=2×0.60152+2×0.37172=1.0 C3:q3=2×0.60152+2×(-0.3717)2=1.0 C4:q4=2×0.37172+2×(-0.6015)2=1.0 (2) 键级:p12=2×0.3717×0.6015+2 × 0.6015×0.3717=0.894p23=2×0.6015×0.6015+2 × 0.3717×(-0.3717)=0.447 p34= p12总键级P12=P34=1.894 P23=1.447 (3) 自由价:F1=4.732-(1+1+1.894)=0.838 F2=4.732-(1+1.894+1.447)=0.391 F3=F2 F4= F1(4) 分子图:例1:休克尔分子轨道近似法处理烯丙基[·CH2-CH =CH2] 的结构。
(1)写出休克尔行列式 (2)解行列式,求出轨道能 (3)计算分子离域能C CH CH CHHH H 123423221HC H C H C =-∙011101=χχχ33∏(1)(2) x 3-2x=02,0±==x x βα21+=E α=2E βα23-=E 432144321343212432113717.06015.06015.03717.06015.03717.03717.06015.06015.03717.03717.06015.03717.06015.06015.03717.0φφφφψφφφφψφφφφψφφφφψ-+-=+--=--+=+++=2221ψψ131221ψψψ例2:用HMO 方法处理三亚甲基甲烷得四个π轨道及相应能量为:(1) 根据能级分布写出电子排布情况(2) 求中心C 原子的键级(3) 求C1-C4上的π电荷密度(1)根据能级分布E2=E3,为简并能级,故电子排布为(2) π键级P12=2⨯0.7071⨯0.4082+1⨯0⨯0.7071+1⨯0⨯0.4082=0.5773 根据对称性可知P13=P14=P12=0.5773 中心碳原子总键级(成键度):N1=3+0.5773⨯3=4.732 (3) π电荷密度q1=2⨯0.70712+1⨯02+ 1⨯02=1.00q2=2⨯0.40822+1⨯0.70712+1⨯0.40822=1.00 q3=q4 =q2=1.0例:下列四种络合物中,d-d 跃迁能量(分裂 能)最低的是( ) a. [Fe(H2O)6] 2+ b. [Fe(H2O)6] 3+ c. [FeF6] 4- d. [FeF6] 3-1. 已知:[Co(CN)6]3-:Δ=34000 cm-1,P=21000 cm-1 [Fe(H2O)6]3+:Δ=13700 cm –1,P=30000 cm-1试确定上述络合物的磁矩,并计算晶体场稳定化能CFSE 。
对结构化学的理解和看法1.引言1.1 概述概述结构化学是一门研究物质的组成、结构和性质之间相互关系的学科。
它通过分析和解释分子和材料的结构,揭示其内部的组织和相互作用,从而帮助我们更好地理解和利用物质世界。
在过去的几十年中,结构化学发展迅速,并在许多领域取得了重要的成就。
通过运用先进的仪器设备和先进的计算方法,结构化学家能够确定和描述分子和材料的三维结构,进而揭示其物理和化学性质。
结构化学不仅仅关注分子和材料的静态结构,还关注它们之间的相互作用和动态变化。
通过研究化学反应的机理和动力学,结构化学为合成新的化合物和材料提供了理论基础,并为设计和改进具有特定性能的功能材料提供了理论指导。
此外,结构化学在药物设计、材料科学、能源研究、环境保护等领域也发挥着重要的作用。
例如,通过研究分子的结构和相互作用,我们能够设计出更有效的药物,开发出更高效的催化剂,探索新型的能源材料,从而推动科学技术的进步和社会的发展。
然而,结构化学仍然面临许多挑战和困扰。
其中之一是如何处理大量的结构数据和复杂的结构关系。
为了更好地理解和利用分子和材料的结构和性质,我们需要开发更加高效和准确的计算方法,并探索更加深入的理论和实验研究。
总的来说,结构化学是一个充满潜力和挑战的学科。
通过深入研究和理解物质的结构和性质,我们可以为科学研究和技术创新提供有力支持,并为解决重大科学问题和社会问题做出重要贡献。
随着科学技术的不断进步,结构化学必将迎来更加广阔的发展前景。
1.2 文章结构本文将分为引言、正文和结论三个部分来展开对结构化学的理解和看法。
在引言部分,将对结构化学进行概述,介绍其基本概念和相关背景信息。
同时,还将对本文的文章结构进行简要说明,以便读者能够更好地理解本文的内容和逻辑。
接下来是正文部分,本部分将分为两个小节来论述结构化学的基本概念和在实际应用中的作用。
在2.1节中,将详细阐述结构化学的基本概念,包括化学键和分子结构等重要内容。
结构化学课程教学设计2019-06-301结构化学的重要性只有让学⽣深刻认识结构化学的重要性,才能使他们产⽣学习兴趣,激发起学习的动⼒,充分发挥其主观能动性,使教学达到事半功倍的效果。
(1)结构化学是化学各学科的理论基础。
结构化学为化学各学科提供理论指导,是联系基础化学与⾼等化学的阶梯。
结构化学已经渗透到现代化学的各个领域。
以学⽣学习过的课程为例,⽆机化学中涉及了原⼦结构、分⼦结构、晶体结构和配合物结构等⽅⾯的内容;有机化学中运⽤杂化轨道理论和分⼦轨道理论说明有机物的结构,使⽤分⼦对称性理论描述分⼦空间结构,利⽤前线轨道理论解释化学反应机理等;仪器分析中紫外光谱中的电⼦跃迁、红外光谱中的简正振动、X射线衍射等,都与结构化学知识紧密相关。
从这些学⽣熟悉的课程⼊⼿,可使他们很快体会到结构化学的重要基础地位。
(2)结构化学是分⼦设计的理论基础。
“结构决定性能,性能反映结构”。
如果找到某类具有特殊性质的物质的规律性,就能设计出性能更好的分⼦。
结构化学及在其基础上发展起来的计算化学、分⼦模拟等对分⼦设计起理论指导作⽤。
为了让学⽣了解这⽅⾯的内容,可⽤如下实例进⾏说明。
⾸先以⽯墨烯为例。
碳元素是⾃然界中分布⼴泛并且与⼈类社会发展关系密切的重要元素。
碳单质有多种存在形式,主要有⽯墨、⾦刚⽯、富勒烯、碳纳⽶管等,其中⽯墨烯由于其优良的结构性质⽽成为材料科学领域的研究热点。
在教学中可先向学⽣提出问题:⽯墨烯的结构是怎样的呢?这就要从⽯墨的结构谈起。
⽯墨为层状结构,同层的碳原⼦间以sp2杂化形成平⾯共价键,每个碳原⼦剩余⼀个p轨道未参与杂化,上⾯各有⼀个电⼦,这些p轨道互相平⾏且与sp2杂化轨道所在平⾯垂直,相互重叠形成离域⼤π键。
π电⼦在整个碳原⼦平⾯⽅向运动,所以⽯墨可以导电和导热,可以⽤来制作电极和坩埚。
⽽⽯墨的层与层之间以微弱的范德华⼒相结合,容易断开⽽滑动,所以⽯墨具有润滑性,可以⽤来制作润滑剂。
⽯墨烯可以看做是只有⼀个原⼦层厚度的单层⽯墨⽚。
《结构化学之计算化学-Gaussian 的操作与练习综述报告》中南大学化学化工学院《结构化学》综述报告结构化学》标题:基于Gauss 03 的操作与练习综述报告指导老师:指导老师:姓学班时名:号:级:间:周德璧******** ********** ******** 2011/1/12 1 《结构化学之计算化学-Gaussian 的操作与练习综述报告》2 《结构化学之计算化学-Gaussian 的操作与练习综述报告》目录简介 (1)一.Gaussian 与GaussView 03 简介(一).关于Gaussian (二). 关于Gaussian 3 (三).GaussView 3 初始界面简介操作实例简介――构建苯乙烷分子 (5)二.操作实例简介操作实例简介说明:在操作的过程中发现,如果仅仅下载周老师在邮箱里面发的GaussView3.07 并进行安装,在进行计算calculate 的操作的时候,总会出现跳出的对话框中的“submit” 选项按钮总是灰色的。
经过上网搜索,网友一致的反应是――没有同时安装相应版本的Gaussian 软件。
因此,我特地下载了Gaussian 03W 软件包,先安装了Gaussian 03W,然后再安装了GaussView3.07。
最后,依据网上查得的指导资料,仿照指导的步骤,亦步亦趋,完成了如下文档。
因软件Gaussian 03W 下载资源很慢,一直到2011/1/12 才下载下来,安装完毕后,因着急要坐火车回家,在构建苯乙烷分子之后仅进行到“Calculation”一步,关于分子结果的可视化的实现没来得及做,请老师谅解。
3 《结构化学之计算化学-Gaussian 的操作与练习综述报告》一. Gaussian 与GaussView 3.07 简介(一).关于Gaussian Gaussian 是一个功能强大的量子化学综合软件包。
其可执行程序可在不同型号的大型计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。
Gaussian 功能主要有:分子能量和结构、过渡态能量和结构、键和反应能量、分子轨道多重矩、原子电荷和电势、振动频率、红外和拉曼光谱、核磁性质、极化率和超极化率、热力学性质、反应路径等。
Gaussian的计算可以对体系的基态或激发态执行。
可以预测周期体系的能量,结构和分子轨道。
因此,Gaussian可以作为功能强大的工具,用于研究许多化学领域的课题,例如取代基的影响,化学反应机理,势能曲面和激发能等等。
(二). 关于Gaussian 03 Gaussian 03 是Gaussian 系列电子结构程序的较为新的版本。
它在化学、化工、生物化学、物理化学等化学相关领域方面的功能都进行了增强。
1.其主要功用大体有以下几个大的方面(1)研究大分子的反应和光谱(2)通过自旋-自旋耦合常数确定构像(3)研究周期性体系(4)预测光谱(5)模拟在反应和分子特性中溶剂的影响2.Gaussian 03 新增加了以下内容:(1).新的量子化学方法(2)新的分子特性(3)新增加的基本算法(4)新增功能:(三).GaussView 3.07 初始界面简介(1)GaussView 是一个专门设计于高斯配套使用的软件,其主要用途有两个构建高斯的输入文件以图的形式显示高斯计算的结果除了可以自己构建输入文件外,GaussView 还可读入CHEM3D,HYPERCHEM 和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围(详见下图)(2)主要功能键的介绍第一行为菜单栏,如下所示: ①File:主要功能是建立,打开,保存和打印当前的文件 4 《结构化学之计算化学-Gaussian 的操作与练习综述报告》Save Image 将当前文件保存为图片格式Preferences。
可以在里面改变Gview 默认的各种显示的设置。
②Edit: 在这里可以完成对分子的剪贴,拷贝,删除和抓图等。
Atom List,显示当前分子的内坐标,笛卡儿坐标,分数坐标等。
Point Group 可以显示当前分子的点群及可能有的点群。
PBC 显示晶体文件(可以将CIF 文件转换为图形,在点PBC 按钮后所给并的对话框中根据选项调节具体显示的格式。
Mos 用于显示分子轨道(只有检查点文件,此选项才能给出分子轨道图。
Symmetrize,对当前体系进行对称性控制。
③View 这里面的选项都是于分子的显示有关的,如显示氢原子,显示键,显示元素符号,显示坐标轴等5 《结构化学之计算化学-Gaussian 的操作与练习综述报告》④Calculate:可从Gview 中直接向高斯提交计算。
这是Gview 作为高斯软件配套功能的重要体现。
从所给的对话框中可以选择工作类型Job Type(如优化,能量或频率等);计算方法Method(如半经验方法,HF 方法,DFT 方法,MP 方法等,还可以选定组);Title(对所要做的计算给一个说明,以备以后的查看) Link 0 (给检查点文件命名,还可以在此用RWF 命令设置临时数据交换文件的大小);General,Guess,(这两个选项主要是给出体系中各原子的连接关系及如何给出初始猜测);NBO(可在此设定NBO 计算),PBC(可在此设定晶体的有关计算), Solvation(可在此设定溶液中的计算,除了选择溶剂外,还要选择模拟溶剂的理论模型)⑤Result :显示计算的结果,包括电荷,静电势表示的表面,振动,频率,核磁,势能面扫描,优化等。
注意有些结果只能用检查点文件才能显示。
6 《结构化学之计算化学-Gaussian 的操作与练习综述报告》7 《结构化学之计算化学-Gaussian 的操作与练习综述报告》二.操作实例简介下面以一个实例介绍一下自己熟悉GaussView 的操作过程。
以以构建一个苯乙烷分子并从Gview 里递交计算为例来说明:构建苯乙烷分子 1.打开Gaussview ,下图就是Gaussview 打开后的窗口2.双击窗口中图标,得到如下窗口里面有常用的环状官能团。
选中苯环(单击即可选中) 3. 在当前工作窗口(打开Gview 时程序自动打开一个工作窗口,如下图)也可通过File-new 路径新建一个工作窗口8 《结构化学之计算化学-Gaussian 的操作与练习综述报告》在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图:将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上,前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子。
当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离可以用Ctrl+Alt+ 鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。
Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调节分子的角度]这个功能连用。
4. 双击Gview 界面上的图标。
出现以下窗口点击氟的元素符号“C”,就选中碳原子。
9 《结构化学之计算化学-Gaussian 的操作与练习综述报告》5. 点击苯环上的任意一个氢原子“H” ,便可得到如下图所示的苯甲烷。
6.在上图所得到的图形中,任选甲基上面任意一个“H”原子,便可得到如下图所示的苯乙烷分子。
同样地,也可以通过类似的操作查看分子的不同角度:将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上,前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子。
当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离可以用Ctrl+Alt+ 鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。
Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调节分子的角度]这个功能连用。
至此,苯乙烷分子的构建都已经完成。
7. Gview 存储个人常用分子的功能。
Gview 中只有常用的一些环状分子和链状分子,远不能10 《结构化学之计算化学-Gaussian 的操作与练习综述报告》满足研究特定体系的特殊需要。
不过Gview 有一个功能可以弥补这个缺憾:可以把常用的分子或官能团存在制定的文件夹内。
在需要时可以直接调用。
双击view 上的相应的图标,在下面的对话框中键入相关项目,保存即可8.查看分子的对称性从Edit-Point group 路径可以查看所构建分子的点群。
点击Point group 后,出现如下窗口:为C1 点群,其下拉菜单中的为可能的点群(改变Tolerance,也可帮助我们判断所构建体系可能有的点群)9.查看分子坐标。
单击Gview 界面上的图标。
出现下面窗口11 《结构化学之计算化学-Gaussian 的操作与练习综述报告》图中:Z 表示时内坐标,C 表示直角坐标。
可以在里面对坐标做适当的调整13.向Gauss 递交计算。
(1)点Gview 界面上Calculation 会出来一个递交计算的对话框。
从所给的对话框中可以选择工作类型Job Type(如优化,能量或频率等);计算方法Method(如半经验方法,HF 方法,DFT 方法,MP 方法等,还可以选定基组);Title(对所要做的计算给一个说明,以备以后的查看) Link 0(给检查点文件命名,还可以在此用RWF 命令设置临时数据交换文件的大小)General, Guess,(这两个选项主要是给出体系中各原子的连接关系及如何给出初;始猜测);NBO(可在此设定NBO 计算),PBC(可在此设定晶体的有关计算), Solvation (可在此设定溶液中的计算,除了选择溶剂外,还要选择模拟溶剂的理论模型)的Gaussian。
在本例中,仅选择如下:Job Type 选项: Hartree-Fock Method 选项:选择如下面的截图所示:然后点击提交“submit” ,可得到对话框如下所示:点击“Save”可得:12 《结构化学之计算化学-Gaussian 的操作与练习综述报告》命名为“苯乙烷” ,然后点击“Save” ,便可得:点击“OK” ,至此,苯乙烷的分子构建的保存已经完成。
(2). 选择完毕后,点Submit 即可递交计算。
有时由于安装的原因Gview 无法与Gauss 建立关联,就不能直接从Gview 里递交计算。
这时可以在Gview 里保存用于Gauss 计算的输入文件,然后从Gauss 里调出文件进行计算。