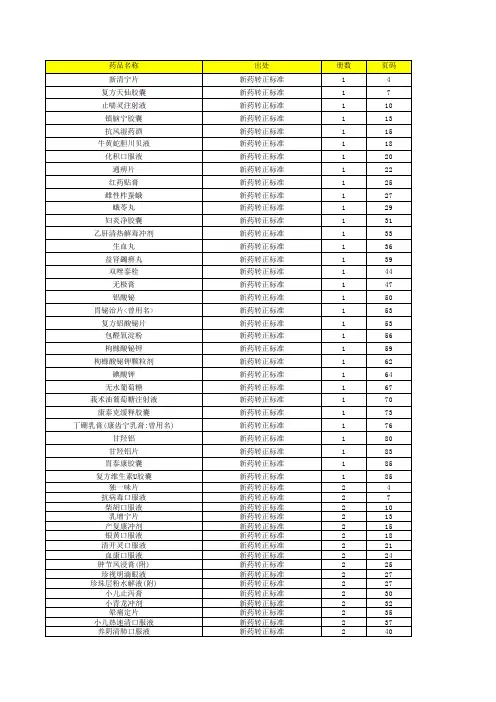
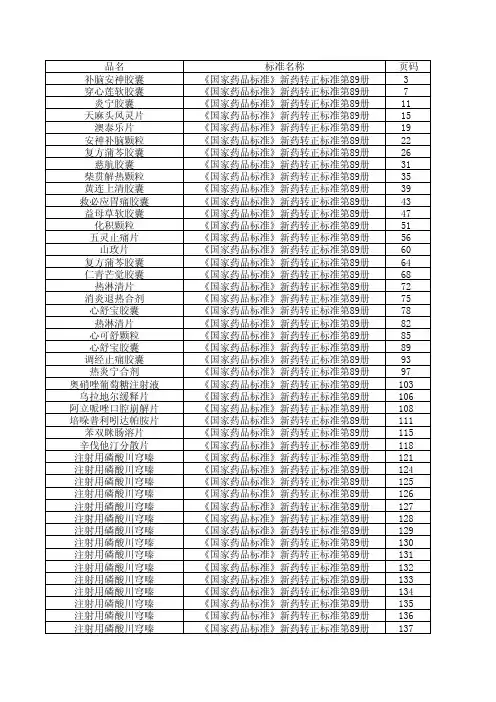
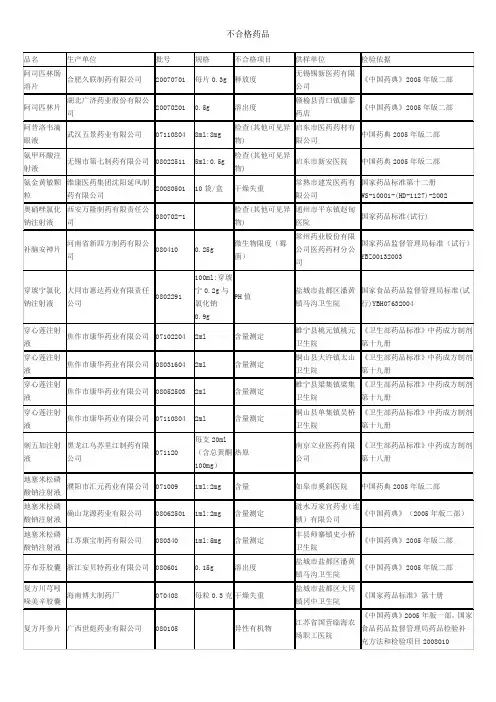
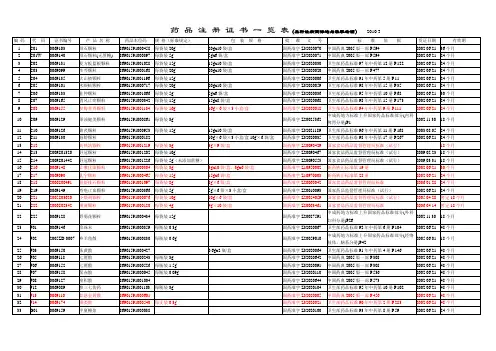
新药转正标准目录1-104册
- 格式:xls
- 大小:532.00 KB
- 文档页数:28

新药转正中药标准卫生部颁药品标准(新药转正标准中药第一册)(15种)雌性柞蚕蛾拼音名:Cixingzuocane英文名:书页号:X1-27 标准编号:WS3-125(Z-31)-92(Z)批准文号:(90)卫药准字Z-19号本品为天蚕蛾科昆虫柞蚕Antheraea Pernyi Guerin-Meneville的未交配的雌性蚕蛾,经排尿、去翅、去足的干燥体。
【性状】本品由头、胸、腹三部分组成。
全身披覆深浅不同的黄褐色短毛。
体长约4cm,头部小,复眼一双,呈深褐色,口器退化,胸部由前胸、中胸、后胸三部组成腹部肥硕,末端较圆,可认出七个环节。
【鉴别】取本品4g,研细,加无水乙醇40ml,振摇5分钟,滤过,取滤液,加无水乙醇稀释至50ml,摇匀,吸取1ml置10ml量瓶中,加无水乙醇稀释至刻度,摇匀后照分光光度法(中国药典1990年版一部附录52页)测定,在269nm波长处有最大吸收。
【检查】水分取本品15g,照水分测定法(中国药典1990年版一部附录30页烘干法)测定。
不得过8%。
【炮制】除去杂质,粉碎成细粉。
【性味与归经】咸、温,归肝、肾经。
【功能与主治】补肾益肾,温脾助胃,宁心安神,调气活血。
用于调整内分泌紊乱。
【用法与用量】2~3g,入丸、散用。
【贮藏】置0~-10℃保存。
蛾苓丸拼音名:Eling Wan英文名:书页号:X1-29 标准编号:WS3-125(Z-31)-92(Z)批准文号:(90)卫药准字Z-19号本品为雌性柞蚕蛾等经加工制成的水丸。
【制法】以上药味,粉碎成细粉,混匀,用水泛丸,制成1000粒,烘干,用煅赭石粉挂衣,硅腊粉打光,即得。
【性状】本品为暗红色的水泛丸,除去外衣,显灰黄色或棕黄色;味甘。
【鉴别】(1) 取本品,置显微镜下观察:不规则分枝状团块无色,遇水合氯醛液溶化,菌丝无色或淡棕色,直径4~6μm,细长,稍弯曲有分枝。
(2) 取本品15粒,研细,加无水乙醇30ml,振摇提取5分钟,滤过,取滤液置50ml量瓶中,加乙醇稀释至刻度,摇匀,精密吸取1.0ml,置10ml量瓶中,加乙醇稀释至刻度,摇匀后照分光光度法(中国药典1990年版一部附录52页)测定,在269nm波长处有最大吸收。

新药转正西药标准卫生部颁药品标准(新药转正标准西药第十五册)12种复方盐酸阿米洛利片拼音名:Fufang Yansuan Amiluoli Pian英文名:Compound Amiloride Hydrochloride Tablets书页号:x15-71 标准编号:WS1?X?220?98Z批准文号:(90)卫药准字X-138号(90)卫药准字X-138-2号本品每片含盐酸阿米洛利C6H8ClN7O?HCl应为2.25~2.75mg;含氢氯噻嗪C7H8ClNO4S2应为22.5~27.5mg。
【处方】盐酸阿米洛利 2.5g 氢氯噻嗪 25g 制成 1000片【性状】本品为类白色片。
【鉴别】在含量测定项下记录的色谱图中,供试品峰的保留时间应与对照品峰的保留时间一致。
【检查】含量均匀度取本品1片,置50ml 量瓶中,照含量测定项下的方法测定,应符合规定中国药典1995年版二部附录Ⅹ E。
溶出度取本品,照溶出度测定法中国药典1995年版二部附录Ⅹ C第一法,以0.1mol/L盐酸溶液900ml为溶剂,转速为每分钟100转,依法操作,经30分钟时,取溶液10ml,滤过,滤液备用。
盐酸阿米洛利取上述溶液,照分光光度法中国药典1995年版二部附录Ⅳ A在365nm的波长处测定吸收度,另取经100℃减压干燥3小时的盐酸阿米洛利对照晶适量,加0.1ml/L盐酸溶液制成每1ml含2.5μg的溶液,同法测定吸收度,计算出每片的溶出量,限度为标示量的80%,应符合规定。
氢氯噻嗪取上述溶液1ml,置10ml量瓶中,加0.1mol/L盐酸溶液稀释至刻度,摇匀,照分光光度法中国药典1995年版二部附录Ⅳ A,在272nm波长处测定吸收度。
另取经105℃干燥1小时的氢氯噻嗪对照品,加0.1mol/L盐酸溶液制成每1ml含2.5μg的溶液;同法测定吸收度,计算出每片的溶出量,限度为标示量的80%,应符合规定。
其他应符合片剂项下有关的各项规定中国药典1995年版二部附录Ⅰ A。

新药转正中药标准 卫生部颁药品标准(新药转正标准中药第二十六册) (16 种)味微苦。
【鉴别】 另取黄芪甲甙对照品, 层色谱法(中国药典1995年版一部附录WB)试验,吸取上述两种溶液各别点于同一硅胶 G 薄层板上,以氯仿-甲醇冰(65:35:10)的下层溶液为展开剂, 展开,取出,晾干,喷以 10%硫酸乙醇溶液,在 105 C 加热约5分钟,置紫外光灯(365nm)下检视。
供试品色谱中,在与对照品色谱相应的位置上,显相同 颜色的荧光斑点。
(2)取本品内容物2g ,加醋酸乙酯30ml ,超声处理10分钟,滤过,滤液置 水浴中浓缩至约1ml ,作为供试品溶液。
另取绿萍对照药材 2g ,同法制成对照药材溶液。
照薄层色谱法(中国药典1995年版一部附录WB)试验,吸取上述两种溶液各10卩I ,分别点于同一硅胶 G 薄层板上,以苯-丙醇(8:1)为展开剂,展开, 取出,晾干,置紫外光灯(365nm)下检视。
供试品色谱中,在与对照药材色谱 相应的位置上,显相同颜色的荧光斑点。
【检查】 应符合胶囊剂项下有关的各项规定 (中国药典 1995年版一部附录I L )。
【含量测定】取本品装量差异项下的内容物2.0g ,精密称定,置索氏提取器中,加 2%氢氧化钠甲醇溶液适量 ,回流提取至近无色,提取液移入100ml烧杯中,用少量甲醇分次洗涤容器,洗液并入烧杯中,置水浴上蒸干,残渣 加水 20ml 使溶解,移至分液漏斗中 ,用氯仿 -正丁醇 (2:1)的混合溶液提取 5次(30、 30、20、20、20ml),合并提取液,用 1 %磷酸二氢钾溶液 50ml 洗涤,弃去水层, 提取液蒸干,残渣加甲醇使溶解,移至 5ml 量瓶中,用甲醇稀释至刻度,作为 供试品溶液。
另取黄芪甲甙对照品,加甲醇制成每 1ml 含 1mg 的溶液,作为对 照品溶液。
照薄层色谱法(中国药典1995年版一部附录W B)试验,精密吸取供 试品溶液5卩I 、对照品溶液2卩I 和4卩I ,分别交叉点于同一硅胶 G 薄层板上,以氯 仿-甲醇-水(65:35:10)的下层液为展开剂,展开,取出,晾干,喷以20%硫酸乙醇溶液,在105 C 加热约5分钟,至斑点显色清晰,取出,在薄层板上覆盖同 样大小的玻璃板,周围用胶布固定,照薄层色谱法 (中国药典 1995年版一部附 录W B 薄层扫描法)进行扫描,波长:入S = 520nm ,入R =700nm ,测量供试品吸 收度积分值与对照品吸收度积分值,计算,即得。

国家药品标准新药转正标准国家药品标准新药转正标准是指新药在经过临床试验并证实其安全性和有效性后,按照国家相关规定转为正式药品的标准。
新药转正标准的制定,对于保障人民群众用药安全、促进药品创新和发展具有重要意义。
首先,国家药品标准新药转正标准应当严格遵循科学、严谨、公正的原则。
在制定新药转正标准时,需要充分考虑药物的药理学、药代动力学、毒理学等方面的科学数据,并结合临床试验结果进行评估和分析,确保新药的安全性和有效性。
同时,还需要充分尊重专业机构和专家的意见,确保标准的科学性和公正性。
其次,国家药品标准新药转正标准应当注重与国际接轨。
随着全球化进程的加快,药品研发和监管已经越来越趋向于国际化。
因此,制定新药转正标准时,需要参考国际上先进的药品标准和规范,借鉴国际经验,促进我国药品标准与国际接轨,提高我国药品的国际竞争力。
再次,国家药品标准新药转正标准应当注重透明和公开。
在制定新药转正标准的过程中,应当建立健全的标准制定程序和机制,确保相关部门和专家的公正、透明和公开,接受社会各界的监督和评价。
同时,还需要建立健全的标准修订机制,及时对新药转正标准进行修订和更新,以适应新药研发和监管的需要。
最后,国家药品标准新药转正标准应当注重风险评估和管理。
在新药转正标准的制定过程中,需要充分考虑新药可能存在的风险和不确定性,建立健全的风险评估和管理体系,制定相应的风险控制措施,确保新药的安全性和有效性。
综上所述,国家药品标准新药转正标准的制定是一个复杂而严谨的过程,需要各方共同努力,确保新药转正标准的科学性、公正性和透明性,促进我国药品创新和发展,保障人民群众用药安全。
希望相关部门和专家能够加强合作,共同推动国家药品标准新药转正标准的制定和完善,为我国药品监管工作提供有力支持。

新药转正标准随着医学技术的不断进步,新药的研发和上市已成为医药行业的重要环节。
然而,新药的研发上市并非易事,需要经历严格的审批和转正标准。
本文将就新药转正标准进行探讨,以期为相关人士提供参考和指导。
首先,新药的转正标准需符合国家相关法律法规的规定。
国家药品监督管理局对新药的转正标准有着明确的规定,包括药物的安全性、有效性、质量控制等方面的要求。
只有符合国家法规的新药才能获得转正资格,才能在市场上合法销售和使用。
其次,新药的转正标准需要经过临床试验的验证。
临床试验是新药研发过程中至关重要的一环,只有通过临床试验并证明其安全有效,新药才能获得转正资格。
临床试验需要进行多阶段、多中心的研究,确保新药在不同人群和不同病情下的安全性和有效性。
除此之外,新药的转正标准还需考虑其与现有药物的比较。
新药需要证明其在疗效、安全性、用药方案等方面与现有药物相比具有明显优势,才能获得转正资格。
这也是为了保障患者的用药安全和疗效,避免新药的上市对患者造成不良影响。
另外,新药的转正标准还需考虑其生产工艺和质量控制。
新药的生产工艺需要符合国家药典的规定,确保药物的质量稳定可控。
同时,新药的质量控制也需要符合相关标准,包括原料药的质量、制剂的稳定性等方面的要求。
总的来说,新药的转正标准是一个综合性的评价体系,需要考虑药物的安全性、有效性、质量控制、临床试验、与现有药物的比较等多个方面。
只有符合这些方面的要求,新药才能获得转正资格,才能在市场上合法上市和销售。
因此,新药的研发和转正标准需要相关人士慎重对待,确保新药的安全有效,为患者的用药提供更多的选择和希望。
希望本文对新药转正标准有所帮助,谢谢阅读。

新药转正标准中药第44册新药转正中药标准卫生部颁药品标准(新药转正标准中药第四十四册)(23种)八珍胶囊拼音名:Bazhen Jiaonang英文名:书页号:X44-73 标准编号:WS3-156(Z-022)-2003(Z)【处方】党参白术(炒) 茯苓甘草当归白芍川芎熟地黄【性状】本品为胶囊剂,内容物为深棕色的颗粒及粉末;气微香,味微甜、苦。
【鉴别】(1)取本品,置显微镜下观察:草酸钙晶体存在于薄壁细胞中,呈类圆形或类簇晶状,直径10,25μm,常数个排列成行。
韧皮薄壁细胞纺锤形,壁略厚,表面有极微细的斜向交错纹理。
(2)取本品内容物2.4g,加乙醇40ml,在水浴上浸渍1小时,时时振摇,滤过,滤液蒸干,残渣加水20ml使溶解,用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇液,用水洗涤3次,每次15ml,弃去水液,正丁醇液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。
另取芍药苷对照品,加乙醇制成每1ml含2mg的溶液,作为对照品溶液。
照薄层色谱法(中国药典2000年版一部附录? B)试验,吸取上述两种溶液各5,10μl,分别点于同一硅胶G薄层板上,以氯仿-醋酸乙酯-甲醇-甲酸(40:5:10:0.2)为展开剂,展开,取出,晾干,喷以5, 香草醛硫酸溶液,加热至斑点显色清晰。
供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
(3)取本品内容物4g,加乙醚60ml,超声处理20分钟,滤过,滤液挥干,残渣加乙醇1ml使溶解,作为供试品溶液。
另取当归、川芎对照药材0.2g,同法制成对照药材溶液。
照薄层色谱法(中国药典2000年版一部附录? B)试验,吸取上述两种溶液各2μl,分别点于同一硅胶H薄层板上,以石油醚(30,60?)-醋酸乙酯-冰醋酸(9:1:0.1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。
供试品色谱中,在与对照药材色谱相应的位置上,分别显相同颜色的荧光斑点。

新药转正西药标准 卫生部颁药品标准(新药转正标准西药第九册) (15 种)对乙酰氨基酚滴剂 拼音名: Duiyixian ※ anjifen Diji 英文名: Paracetamol Drops 书页号: X9-60 批准文号:( 92)卫药准字 X-76 号本品含对乙酰氨基酚(C3H9NO2)应为标示量的90.0〜110.0%。
【性状】 本品为淡红色或红色的澄清液体;臭芳香,味微苦,可加 适宜调味剂。
【鉴别】 在含量测定项下记录的色谱图中,供试品峰的保留时间应 与对照品峰的保留时间一致。
【检查】 对氨基酚 取本品,加水制成每 1ml 中含对乙酰氨基酚 2.0mg 的供试品溶液;另取对氨基酚对照品,加水制成每1ml 中含10卩g 的对照品溶液。
照含量测定项下的色谱条件,用外标法 (中国药典1995年版二部附录V D )测定,精密量取上述两种溶液各 10卩I,分别注入液相色谱仪,记录色谱图。
供试品溶液的色谱图中如出现与对照品溶液相应的对氨基酚峰,其峰面积 不得大于对照品溶液主成分峰的峰面积(0.5% )。
相对密度本品的相对密度(中国药典1995年版二部附录W A )为1.070〜1.150。
pH 值 应为4.7〜7.0(中国药典1995年版二部附录W H )。
装量 取本品,依法检查(中国药典1995年版二部附录XF ),应符合 规定。
【含量测定】 照高效液相色谱法(中国药典1995年版二部附录VD ) 测定。
色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂,甲醇-0.05mol/L 醋酸铵溶液(15:85)为流动相,检测波长为 257nm 。
理论板数按对乙酰氨基酚峰计算应不低于 5000,对乙酰氨基酚峰与内标物质峰的分离度应 符合要求。
内标溶液的制备 取茶碱,加水制成每 1ml 中含 1.0mg 的溶液,摇匀,即 得。
测定法 精密量取本品适量,加水定量稀释制成每 1ml 中约含对乙酰氨 基酚0.6mg 的溶液,精密量取此溶液与内标溶液各 5ml ,置50ml 量瓶中,用水稀释至刻度,摇匀,取 10卩l 注入液相色谱仪,记录色谱图;另精密称取在 105 C干燥至恒重的对乙酰氨基酚对照品适量,加水溶解并定量稀释制成每1ml 中 约含 0.6mg 的溶液,同法测定。
新药转正中药标准卫生部颁药品标准(新药转正标准中药第十一册)(15种)阿胶补血口服液拼音名:Ejiao Buxue Koufuye英文名:书页号:X11-20 标准编号:WS3-74(Z-64)-96(Z)批准文号:(92)卫药准字Z-22号【处方】阿胶熟地黄党参黄芪枸杞子白术【性状】本品为深棕色液体;味微甜。
【鉴别】取本品5ml,加水15ml,搅匀,滤过,取滤液2ml,加茚三酮试液2滴,置沸水浴中加热5分钟,显紫红色。
【检查】相对密度应不低于1.05(中国药典1995年版一部附录ⅦA)。
pH值应为4.0~6.0(中国药典1995年版一部附录ⅦG)。
其他应符合合剂项下有关的各项规定(中国药典1995年版一部附录ⅠJ)。
【含量测定】精密量取本品0.2ml,照氮测定法(中国药典1995年版一部附录ⅨL第二法)测定,总氮量应不少于7.0mg/ml。
【功能与主治】滋阴补血,补中益气,健脾润肺。
用于久病体弱,血亏目眩,虚痨咳嗽。
【用法与用量】口服,一次20ml,早晚各一次或遵医嘱。
【规格】每支20ml【贮藏】密封,置阴凉处。
【使用期限】2年。
阿胶颗粒拼音名:Ejiao Keli英文名:书页号:X11-11 标准编号:WS3-88(Z-13)-96(Z)批准文号:(92)卫药准字Z-01号本品为阿胶经干燥、粉碎,制成的颗粒。
【性状】本品为棕色颗粒或粉末;气香,味微甘。
【检查】水分取本品1g,精密称定,照水分测定法(中国药典1995年版一部附录ⅨH烘干法)测定,不得过7.0%。
总灰分取本品1g,精密称定,照灰分测定法(中国药典1995年版一部附录ⅨK)测定,不得过1.1%。
重金属砷盐应符合阿胶项下有关的各项规定(中国药典1995年版一部513页)。
挥发性碱性物质精密称取本品5g,置100ml量瓶中,加水使溶解并稀释至刻度,摇匀,精密量取5ml置凯式蒸馏瓶中,立刻加1%氢氧化镁混悬溶液5ml,迅速密塞,通入水蒸气进行蒸馏,以2%硼酸溶液5ml为接收液,加甲基红-溴甲酚绿混合指示液5滴,从滴出第一滴凝结水珠时起,蒸馏7分钟停止,馏出液照氮测定法(中国药典1995年版一部附录ⅨL第二法)滴定,即得。
新药转正标准
新药转正标准是指新药从临床试验阶段成功转化为市场上销售的标准。
新药的研发和上市是一个复杂而又严格的过程,其中新药转正标准是至关重要的一环。
本文将从药物临床试验、药物质量标准、药物生产工艺等几个方面来探讨新药转正标准的相关内容。
首先,药物临床试验是新药转正的重要环节。
临床试验是指在人体上进行的药物试验,其目的是评价药物的安全性和有效性。
在临床试验阶段,药物需要经历临床前研究、临床试验设计、临床试验执行和临床试验报告等多个环节。
只有经过严格的临床试验,并获得相关部门的批准,药物才能进入下一阶段的生产制造。
其次,药物质量标准也是新药转正的重要考量因素。
药物质量标准是指药物在生产过程中应符合的一系列标准和规定,包括药物的纯度、稳定性、溶解度等多个方面。
只有符合相关的药物质量标准,药物才能保证在生产和使用过程中的质量和安全。
此外,药物生产工艺也是新药转正的关键环节之一。
药物生产工艺是指药物从原料到成品的整个生产过程,包括原料的采购、生产工艺的设计、生产设备的选择等多个方面。
在药物生产工艺中,需要严格按照相关的法规和标准进行操作,确保药物的生产过程符合规范,从而保证药物的质量和安全。
综上所述,新药转正标准涉及到药物临床试验、药物质量标准、药物生产工艺等多个方面,是一个综合性的考量。
只有在这些方面都符合相关的标准和规定,新药才能成功转正并投放市场。
因此,对于新药的研发和上市,需要严格遵循相关的法规和标准,确保新药的质量和安全,为人们的健康提供保障。
根据药智网药品标准在线查询数据库整理1.中国药典中国药典2010年版一部中国药典2010年版二部中国药典2010年版三部中国药典2010年版一部勘误表中国药典2010年版二部勘误表中国药典2010年版三部勘误表中国药典2005年版一部中国药典2005年版二部中国药典2005年版三部中国药典2005版勘误一部中国药典2005版勘误二部2005药典增补本(2006)二部2005年药典增补(2009)中国药典2000年版一部中国药典2000年版二部2000年版2002年增补本2000年版2004年增补本2.国监局单页标准国监局单页标准(2000年前)国监局单页标准(2000)国监局单页标准(2001)国监局单页标准(2002)国监局单页标准(2003)国监局单页标准(2004)国监局单页标准(2005)国监局单页标准(2006)国监局单页标准(2007)国监局单页标准(2008)国监局单页标准(2009)国监局单页标准(2010)药典委员会勘误国家药品标准修订件药品检验补充检验方法和项目化学药品地标升国标16册化学药品地标升国标16册(修订)国药典综发(2006)3.中成药标准汇编 (地标升国标)内科肾系分册内科心系分册内科肺系分册骨伤科分册口腔肿瘤儿科分册内科肝胆分册内科脾胃分册内科气血津液分册脑系经络肢体分册外科妇科分册眼科耳鼻喉科皮肤科分册4.进口药品标准进口药复核标准汇编(2003)进口药品复核标准汇编2001年进口药品复核标准汇编2000年进口药品复核标准汇编2002年进口药品单页标准(2003)进口药品单页标准(2004)进口药品单页标准(2005)进口药品单页标准(2006)进口药品单页标准(2007)5.卫生部药品标准卫生部药品标准(二部)第一册卫生部药品标准(二部)第二册卫生部药品标准(二部)第三册卫生部药品标准(二部)第四册卫生部药品标准(二部)第五册卫生部药品标准(二部)第六册生化药品89年版第一册抗生素药品第一册<八九年>化学药品及制剂第一册藏药第一册蒙药分册维吾尔药分册6.中药成方制剂第一册第二册第三册第四册第五册第六册第七册第八册第九册第十册第十一册第十二册第十三册第十四册第十五册第十六册第十七册第十八册第十九册第二十册第二十一册(中药保护) 7.新药转正标准新药转正标准1新药转正标准2新药转正标准3新药转正标准4新药转正标准5新药转正标准6新药转正标准7新药转正标准8新药转正标准9新药转正标准10新药转正标准11新药转正标准12新药转正标准13新药转正标准14新药转正标准15新药转正标准16新药转正标准17新药转正标准18新药转正标准19新药转正标准20新药转正标准21新药转正标准22新药转正标准23新药转正标准24新药转正标准25新药转正标准26新药转正标准27新药转正标准28新药转正标准29新药转正标准30新药转正标准31新药转正标准32新药转正标准33新药转正标准34新药转正标准35新药转正标准36新药转正标准37新药转正标准38新药转正标准39新药转正标准40新药转正标准41新药转正标准42新药转正标准43新药转正标准44新药转正标准45新药转正标准46新药转正标准47新药转正标准48新药转正标准49新药转正标准50新药转正标准51新药转正标准52新药转正标准53新药转正标准54新药转正标准55新药转正标准56新药转正标准57新药转正标准58新药转正标准59新药转正标准60新药转正标准61新药转正标准62新药转正标准63新药转正标准64新药转正标准65新药转正标准66新药转正标准67新药转正标准68新药转正标准69新药转正标准70新药转正标准71新药转正标准72新药转正标准73新药转正标准74新药转正标准75新药转正标准768.化药地标升国标化学药品地标升国标1册化学药品地标升国标2册化学药品地标升国标3册化学药品地标升国标4册化学药品地标升国标5册化学药品地标升国标6册化学药品地标升国标7册化学药品地标升国标8册化学药品地标升国标9册化学药品地标升国标10册化学药品地标升国标11册化学药品地标升国标12册化学药品地标升国标13册化学药品地标升国标14册化学药品地标升国标15册化学药品地标升国标16册。
新药转正中药标准卫生部颁药品标准(新药转正标准中药第十二册)(14种)肝达康片拼音名:Gandakang Pian英文名:书页号:X12-23 标准编号:WS3-048(Z-007)-96(Z)批准文号:(94)卫药准字Z-27号【处方】北柴胡(醋炙)白芍(醋炙) 当归(酒炙) 茜草白术(麸炒) 茯苓鳖甲(醋炙) 湘曲党参白茅根枳实(麸炒) 青皮(麸炒)砂仁地龙(炒) 甘草【性状】本品为薄膜衣片,除去薄膜衣后呈棕褐色或黑褐色;气香,味微苦咸。
【鉴别】(1)取本品10片,研细,置挥发油提取器中,从刻度管加水70ml、石油醚(60~90℃)2ml,蒸馏30分钟,分取石油醚层,挥干,残渣滴加5%香草醛硫酸溶液数滴, 即显紫堇色。
(2)取本品20片,除去包衣,研细,加甲醇30ml,超声处理30分钟,滤过,滤液加在预先处理好的中性氧化铝柱(100~200目,5g,内径10~15mm)上,用40%甲醇溶液100ml洗脱,收集洗脱液,置水浴上蒸干,残渣加水15ml使溶解,用水饱和的正丁醇振摇提取3次,每次15ml,合并正丁醇液,用水洗涤2次,每次15ml,弃去水液,取正丁醇液置水浴上蒸干,残渣加甲醇2ml使溶解。
另取芍药甙对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。
照薄层色谱法(中国药典1995年版一部附录ⅥB)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以氯仿-甲醇-甲酸(8:2:0.1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,热风吹至斑点显色清晰。
供试品色谱中, 在与对照品色谱相应的位置上,显相同的蓝紫色斑点。
(3)取北柴胡对照药材1g,加甲醇10ml,超声处理30分钟,滤过,滤液置水浴上蒸干,残渣加甲醇2ml使溶解,作为对照药材溶液。
照薄层色谱法(中国药典1995年版一部附录ⅥB)试验,吸取[鉴别](2)项下的供试品溶液与上述对照药材溶液各2μl,分别点于同一硅胶G薄层板上,以醋酸乙酯-乙醇-水(18:2:1)为展开剂,展开,取出,晾干,喷以1%对二甲氨基苯甲醛乙醇溶液-10%硫酸溶液(1:1)的混合液,在105℃烘烤数分钟。