金属有机反应 (1)
- 格式:ppt
- 大小:2.04 MB
- 文档页数:77


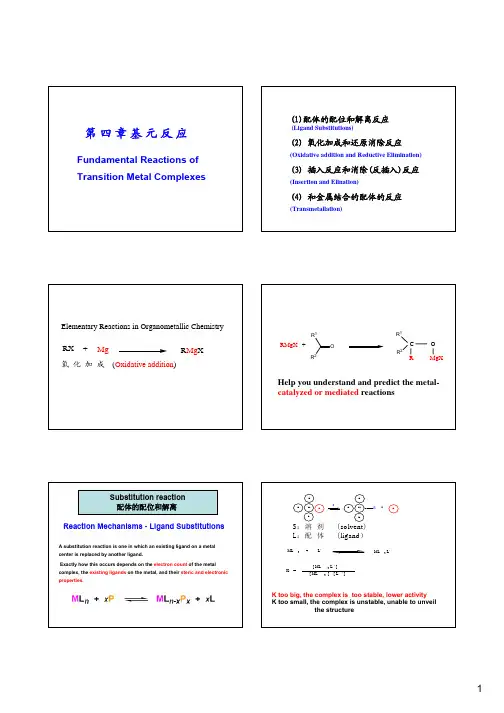
A ligand substitution can occur either by an associativeor dissociative route.In a dissociative substitution, one of the existing ligandson the metal center has to fall off (ligand dissociation),LnM+ :L'' --------> LnM-L"fastthis opens up a free coordination site to which the newligand can coordinate.S‡is significantly positive, the mechanism is almost certainlydissociative.If the rate does not depend on [L], the mechanism is almost certainlydissociative.axial site.The 14e-three coordinate intermediate is actually almost immediately coordinated by a solvent molecule to produce theConsider the following two substitution reactions.the most stable productSince we are starting with an 18e-complex, we have to proceed by adissociative substitution reaction. The phosphite ligand is the more weaklycoordinated and somewhat bulky due to the t-butyl groups. Thus it is the mostlikely ligand to fall off first.The P(OMe)3ligand has about the same σ-donorability as pyridine, but is a considerably better acceptor ligand, thus completing with the trans* Rate depends on both LnM-L' and L"ΔS‡is significantly negative, the mechanism is almost certainlyAssociative SubstitutionsThese occur first by a ligand addition to the metal complexfollowed by the dissociation of one of the original ligands.typically need to have an unsaturated(17e-or lower) complex in order topropose an associative substitution mechanism.there are very few verified examples of this in theliterature.Since we are starting with an 16e-Ni complex without any stericproblems we can to proceed by an associative substitutionreaction. Once the CO ligand coordinates, the weakest ligandthat should fall off is the THF. Late transition metals have a farPentadienylThe pentadienyl ligand is an acyclic version of Cp that does not have any aromatic stabilization. This has two important effects:Consider the following two reactions:Reaction a) is much faster than reaction b). Discuss why this is so.NitrosylThis can occasionally lead to interesting behavior where the linearIn general the stereochemistry of a square-planarcomplex is retained through the substitution reaction.。
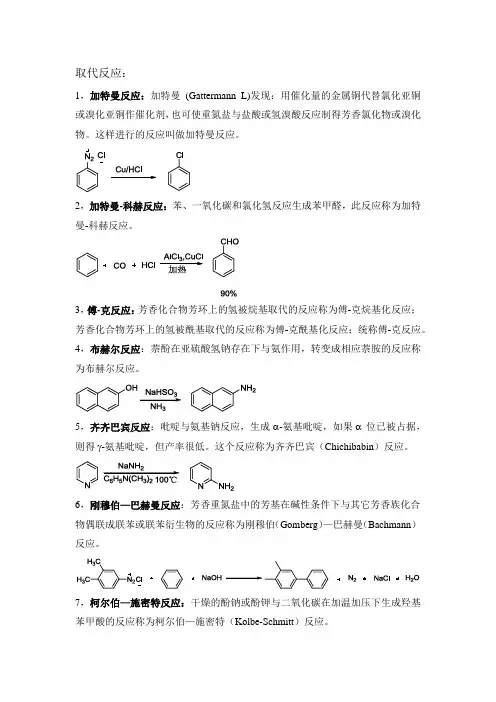
取代反应:1,加特曼反应:加特曼(Gattermann L)发现:用催化量的金属铜代替氯化亚铜或溴化亚铜作催化剂,也可使重氮盐与盐酸或氢溴酸反应制得芳香氯化物或溴化物。
这样进行的反应叫做加特曼反应。
2,加特曼-科赫反应:苯、一氧化碳和氯化氢反应生成苯甲醛,此反应称为加特曼-科赫反应。
3,傅-克反应:芳香化合物芳环上的氢被烷基取代的反应称为傅-克烷基化反应;芳香化合物芳环上的氢被酰基取代的反应称为傅-克酰基化反应;统称傅-克反应。
4,布赫尔反应:萘酚在亚硫酸氢钠存在下与氨作用,转变成相应萘胺的反应称为布赫尔反应。
5,齐齐巴宾反应:吡啶与氨基钠反应,生成α-氨基吡啶,如果α位已被占据,则得γ-氨基吡啶,但产率很低。
这个反应称为齐齐巴宾(Chichibabin)反应。
6,刚穆伯—巴赫曼反应:芳香重氮盐中的芳基在碱性条件下与其它芳香族化合物偶联成联苯或联苯衍生物的反应称为刚穆伯(Gomberg)—巴赫曼(Bachmann)反应。
7,柯尔伯—施密特反应:干燥的酚钠或酚钾与二氧化碳在加温加压下生成羟基苯甲酸的反应称为柯尔伯—施密特(Kolbe-Schmitt)反应。
8,威廉森合成法:在无水条件下,醇钠和卤代烷作用生成醚的反应称为威廉森(Williamson A W)合成法。
9,席曼反应:芳香重氮盐和氟硼酸反应,生成溶解度较小的氟硼酸盐,后者加热分解产生氟苯,这称为席曼(Schiemann)反应。
10,桑德迈耳反应:1884年,桑德迈耳(Sandmeyer T)发现:在氯化亚铜或溴化亚铜的催化下,重氮盐在氢卤酸溶液中加热,重氮基可分别被氯或溴原子取代,生成芳香氯化物或溴化物。
这一反应称为桑德迈耳反应。
11,普塑尔反应:一些重氮盐在碱性条件下或稀酸的条件下可以发生分子内的偶联反应。
这个反应是普塑尔(Pschorr R)在寻找合成菲环的新方法中首先发现的,故称为普塑尔反应。
12,瑞穆尔—悌曼反应:酚与氯仿在碱性溶液中加热生成邻位及对位羟基醛的反应称为瑞穆尔—悌曼(Reimer —Tiemann)反应。

A ligand substitution can occur either by an associativeor dissociative route.In a dissociative substitution, one of the existing ligandson the metal center has to fall off (ligand dissociation),LnM+ :L'' --------> LnM-L"fastthis opens up a free coordination site to which the newligand can coordinate.S‡is significantly positive, the mechanism is almost certainlydissociative.If the rate does not depend on [L], the mechanism is almost certainlydissociative.axial site.The 14e-three coordinate intermediate is actually almost immediately coordinated by a solvent molecule to produce theConsider the following two substitution reactions.the most stable productSince we are starting with an 18e-complex, we have to proceed by adissociative substitution reaction. The phosphite ligand is the more weaklycoordinated and somewhat bulky due to the t-butyl groups. Thus it is the mostlikely ligand to fall off first.The P(OMe)3ligand has about the same σ-donorability as pyridine, but is a considerably better acceptor ligand, thus completing with the trans* Rate depends on both LnM-L' and L"ΔS‡is significantly negative, the mechanism is almost certainlyAssociative SubstitutionsThese occur first by a ligand addition to the metal complexfollowed by the dissociation of one of the original ligands.typically need to have an unsaturated(17e-or lower) complex in order topropose an associative substitution mechanism.there are very few verified examples of this in theliterature.Since we are starting with an 16e-Ni complex without any stericproblems we can to proceed by an associative substitutionreaction. Once the CO ligand coordinates, the weakest ligandthat should fall off is the THF. Late transition metals have a farPentadienylThe pentadienyl ligand is an acyclic version of Cp that does not have any aromatic stabilization. This has two important effects:Consider the following two reactions:Reaction a) is much faster than reaction b). Discuss why this is so.NitrosylThis can occasionally lead to interesting behavior where the linearIn general the stereochemistry of a square-planarcomplex is retained through the substitution reaction.。

基础有机化学反应总结一、烯烃1、卤化氢加成(1)【马氏规则】在不对称烯烃加成中,氢总是加在含碳较多的碳上。
【机理】【本质】不对称烯烃的亲电加成总是生成较稳定的碳正离子中间体。
【注】碳正离子的重排(2)【特点】反马氏规则【机理】自由基机理(略)【注】过氧化物效应仅限于HBr、对HCl、HI无效。
【本质】不对称烯烃加成时生成稳定的自由基中间体。
【例】2、硼氢化—氧化【特点】不对称烯烃经硼氢化—氧化得一反马氏加成的醇,加成是顺式的,并且不重排。
【机理】【例】3、X2加成【机理】【注】通过机理可以看出,反应先形成三元环的溴鎓正离子,然后亲和试剂进攻从背面进攻,不难看出是反式加成。
不对称的烯烃,亲核试剂进攻主要取决于空间效应。
【特点】反式加成4、烯烃的氧化1)稀冷高锰酸钾氧化成邻二醇。
3H 33H3稀冷KMnO 433M nO OOO H 2O 3H 33H 3 2)热浓酸性高锰酸钾氧化3)臭氧氧化4)过氧酸氧化5、烯烃的复分解反应【例】6、共轭二烯烃1)卤化氢加成2)狄尔斯-阿德尔(Diels-Alder )反应【描述】共轭二烯烃和烯烃在加热的条件下很容易生成环状的1,4加成产物。
【例】二、脂环烃1、环丙烷的化学反应【描述】三元环由于张力而不稳定,易发生加成反应开环,类似碳碳双键。
【特点】环烷烃都有抗氧化性,可用于区分不饱和化合物。
【注】遵循马氏规则【例】2、环烷烃制备1)武兹(Wurtz)反应【描述】通过碱金属脱去卤素,制备环烷烃。
【例】2)卡宾①卡宾的生成A、多卤代物的α消除B、由某些双键化合物的分解②卡宾及烯烃的加成反应【特点】顺式加成,构型保持【例】③类卡宾【描述】类卡宾是一类在反应中能起到卡宾作用的非卡宾类化合物,最常用的类卡宾是ICH2ZnI。
【特点】顺式加成,构型保持【例】三、炔烃1、还原成烯烃1)、顺式加成2)、反式加成2、亲电加成1)、加X 2【机理】中间体Br+R 2R 1【特点】反式加成 2)、加HXR R HBr RR Br H (一摩尔的卤化氢主要为反式加成)3)、加H 2O【机理】【特点】炔烃水合符合马式规则。



有机化学人名反应取代反应:1,加特曼反应:加特曼(GattermannL)发现:用催化量的金属铜代替氯化亚铜或溴化亚铜作催化剂,也可使重氮盐与盐酸或氢溴酸反应制得芳香氯化物或溴化物。
这样进行的反应叫做加特曼反应。
2,加特曼-科赫反应:苯、一氧化碳和氯化氢反应生成苯甲醛,此反应称为加特曼-科赫反应。
3,傅-克反应:芳香化合物芳环上的氢被烷基取代的反应称为傅-克烷基化反应;芳香化合物芳环上的氢被酰基取代的反应称为傅-克酰基化反应;统称傅-克反应。
4,布赫尔反应:萘酚在亚硫酸氢钠存在下与氨作用,转变成相应萘胺的反应称为布赫尔反应。
5,齐齐巴宾反应:吡啶与氨基钠反应,生成-氨基吡啶,如果位已被占据,则得-氨基吡啶,但产率很低。
这个反应称为齐齐巴宾(Chichibabin)反应。
6,刚穆伯—巴赫曼反应:芳香重氮盐中的芳基在碱性条件下与其它芳香族化合物偶联成联苯或联苯衍生物的反应称为刚穆伯(Gomberg)—巴赫曼(Bachmann)反应。
7,柯尔伯—施密特反应:干燥的酚钠或酚钾与二氧化碳在加温加压下生成羟基苯甲酸的反应称为柯尔伯—施密特(Kolbe-Schmitt)反应。
8,威廉森合成法:在无水条件下,醇钠和卤代烷作用生成醚的反应称为威廉森(WilliamonAW)合成法。
9,席曼反应:芳香重氮盐和氟硼酸反应,生成溶解度较小的氟硼酸盐,后者加热分解产生氟苯,这称为席曼(Schiemann)反应。
10,桑德迈耳反应:1884年,桑德迈耳(SandmeyerT)发现:在氯化亚铜或溴化亚铜的催化下,重氮盐在氢卤酸溶液中加热,重氮基可分别被氯或溴原子取代,生成芳香氯化物或溴化物。
这一反应称为桑德迈耳反应。
11,普塑尔反应:一些重氮盐在碱性条件下或稀酸的条件下可以发生分子内的偶联反应。
这个反应是普塑尔(PchorrR)在寻找合成菲环的新方法中首先发现的,故称为普塑尔反应。
12,瑞穆尔—悌曼反应:酚与氯仿在碱性溶液中加热生成邻位及对位羟基醛的反应称为瑞穆尔—悌曼(Reimer—Tiemann)反应。
什么是金属有机化学(一)引言概述:金属有机化学是研究金属与有机化合物相互作用和反应机理的学科,它是无机化学和有机化学的交叉领域。
本文将从金属有机化学的定义、发展历程、主要研究对象、研究方法和应用领域等五个大点进行阐述。
正文内容:一、定义1. 金属有机化学的基本概念2. 金属有机化合物的特点和性质3. 金属有机配合物的结构和命名规则4. 金属有机化学与有机化学、无机化学的联系和区别5. 金属有机化学的学科发展意义二、发展历程1. 金属有机化学的起源和发展背景2. 金属有机化学的里程碑事件和重要贡献者3. 金属有机化学在有机合成和无机材料领域的应用突破4. 金属有机化学的前沿研究方向和趋势5. 金属有机化学在实际应用中的发展状况及前景三、主要研究对象1. 金属有机配合物的合成方法和策略2. 金属有机配合物的结构和性质表征技术3. 金属有机配合物的反应机理和动力学研究4. 金属有机配合物的催化应用和机理探究5. 金属有机配合物的生物医学和材料科学应用研究四、研究方法1. 基于有机合成的金属有机化学研究方法2. 基于无机配位化学的金属有机化学研究方法3. 基于物理化学和表面化学的金属有机化学研究方法4. 基于光谱技术的金属有机化学研究方法5. 基于计算化学的金属有机化学研究方法五、应用领域1. 金属有机化学在有机合成中的应用2. 金属有机化学在药物研发中的应用3. 金属有机化学在催化反应中的应用4. 金属有机化学在材料科学中的应用5. 金属有机化学在能源领域中的应用总结:金属有机化学作为一个重要的交叉学科,深入研究金属与有机化合物之间的相互作用和反应机理,对于推动科学和技术的发展具有重要的意义。
随着研究方法的不断创新以及应用领域的拓展,金属有机化学将在有机合成、药物研发、催化反应、材料科学和能源领域等方面发挥越来越大的作用。
有机化学反应历程1四.有机反应历程1.有机反应类型(1)加成反应亲电加成 C=C,C≡C;亲核加成 C=O, C≡C,C≡N;带有吸电⼦基团的加成C=C,如C=C-C=O,C=C-C≡N;⾃由基加成C=C。
(2)取代反应取代反应有三种:亲电取代,重要的是芳环上H 被取代;亲核取代,经常是⾮H原⼦被取代;⾃由基取代,重要的是α取代。
(3)消除反应主要是1,2-消除⽣成烯,也有1,1-消除⽣成碳烯。
(4)重排反应常见的是碳正离⼦重排或其它缺电⼦的中间体重排。
(5)周环反应包括电环化反应、环加成反应及σ迁移反应。
2.反应活性中间体主要活性中间体有其它活性中间体有碳烯R2C∶(卡宾Carbene)氮烯RN∶(乃春 Nitrene);苯炔(Benzyne)。
(1)⾃由基⾃由基的相对稳定性可以从C—H键离解能⼤⼩判别,键离解能越⼤,⾃由基稳定性越⼩。
如按稳定性次序排列R3C·>R2CH·>RCH2·>CH3·C—H键离解能(kJ/mol):380.7 395.4 410.0 435.1C6H5CH2·≈CH2=CH-CH2·>R3C·C—H键离解能(kJ/mol):355.6 355.5Ph3C·>Ph2CH·>PhCH2·Ph3C·为涡轮形,具有约30°夹⾓,因此稳定性不会⽐Ph2CH·⾼得很多,且易发⽣⼆聚形成酿式结构。
【例1】下列游离基哪⼀个最稳定?B.CH2=CHCH2· D.CH3·解:B。
(2)碳正离⼦含有带正电荷的三价碳原⼦的化合物叫碳正离⼦,它具有6个价电⼦,⼀般情况下是sp2杂化,平⾯构型,其稳定性次序为:任何使正电荷离域的条件都能稳定碳正离⼦。
孤电⼦对能分散正电荷故MeOCH2Cl溶剂解反应⽐CH3Cl快1014倍。
邻基效应⽣成桥式碳正离⼦芳⾹化稳定碳正离⼦,例如(3)碳负离⼦碳负离⼦是碳原⼦上带有负电荷的体系,其结构⼤多是⾓锥形sp3杂化构型,此构型使孤电⼦对和三对成键电⼦之间相斥作⽤最⼩。
一种高收率uio-66金属有机框架材料的制备方法及应用UIO-66金属有机框架材料是一种具有多孔性和高表面积的材料,广泛应用于催化、气体吸附和分离等领域。
本文将介绍一种高收率的UIO-66金属有机框架材料的制备方法,并探讨其在催化反应中的应用。
一、制备方法:1.1 原材料准备:首先,准备UIO-66金属有机框架材料的制备所需的原材料,包括金属离子、有机配体、溶剂等。
1.2 合成步骤:a) 在一个干燥的反应器中,加入适量的金属离子盐和有机配体,并加入适量的溶剂,形成反应混合物。
b) 将反应混合物进行搅拌,并控制反应温度和反应时间,使反应物充分反应。
c) 反应结束后,用适量的溶剂洗涤产物,将产物分离出来。
d) 最后,将分离得到的UIO-66金属有机框架材料进行干燥,得到纯净的产物。
1.3 优化工艺:为了获得高收率的UIO-66金属有机框架材料,可以对制备过程中的反应温度、反应时间、溶剂种类和比例等参数进行优化,以提高产物的收率和质量。
UIO-66金属有机框架材料在催化反应中具有广泛的应用,以下是几个典型的应用示例:2.1 催化剂:由于UIO-66金属有机框架材料具有高度可控的孔结构和丰富的活性位点,可以作为催化剂催化各种重要有机反应。
例如,可以将某种金属离子掺杂到UIO-66金属有机框架材料中,形成金属有机框架催化剂,并用于催化氧化反应、还原反应等。
2.2 气体吸附和分离:由于UIO-66金属有机框架材料具有高度可调控的孔结构和大的表面积,可以用于气体的吸附和分离。
例如,可以利用UIO-66金属有机框架材料去除废气中的有害气体,或者用于分离气体混合物中的成分。
2.3 药物输送:UIO-66金属有机框架材料还可以用作药物的载体,实现药物的控释和靶向输送。
通过调整UIO-66金属有机框架材料的孔径和表面性质,可以将药物吸附在孔隙中,并实现药物的缓慢释放,提高药物的疗效。
通过对UIO-66金属有机框架材料的制备方法及应用的研究,我们可以得出结论,使用合适的原材料和制备工艺,可以制备出高收率的UIO-66金属有机框架材料,并将其应用于催化、气体吸附和分离以及药物输送等领域,具有广阔的发展前景。