western blot protocol from HT
- 格式:doc
- 大小:34.50 KB
- 文档页数:4

Western Bloting (张雪雁)操作步骤:1.做胶:下层胶7.5ml一块,以水或乙醇隔离空气。
待下层胶凝固后做上层胶,每块胶配置3ml,灌胶、插梳子,赶走气泡。
2.蛋白变性:在胶凝固过程中,变性蛋白质,蛋白质的上样量按事先所测弄浓度计算准确,分装到EP管或,按5ul/100ul样品的浓度加β—巯基乙醇,然后以PCR仪95℃变性10分钟。
3.上样、电泳:1)上层胶为安全凝固(大约15分钟)后,两只手同时向上用力取下梳子。
以蒸馏水仔细清洗各泳道内残留得碎胶。
2)将SDS—PAGE胶转移到电泳槽中,如果只做一块胶,对面以玻璃板平衡。
1х电泳缓冲液充满电泳槽后,以玻璃棒将电泳槽中的气泡尽量赶除。
3)先以少量加有溴酚蓝1х上样缓冲液标记各泳道。
4)变性过的各管蛋白质样品以加有溴酚蓝1х上样缓冲液补齐至50ul。
蛋白Marker也补齐至50ul。
5)以一定顺序将蛋白样品和Marker,用微量移液器加至泳道。
上样过程中,手要稳,避免将泳道划破,样品自枪头打出时要缓慢,以免样品自泳道飘出。
剩余未加样的泳道以50ul上样缓冲液平衡。
6)加样完毕,红对红,黑对黑将电泳槽和电泳仪接好,打开电源,电压调至60-100V之间。
一般来说样品在上层胶时电压可稍高,进入下层胶后较低的电压容易将个分子量蛋白粉理清楚。
溴酚蓝自胶内跑出后电泳结束。
4.电转:1)准备PVDF膜及滤纸。
PVDF大小8.2х5~5.5,滤纸按电转海绵大小剪,一块胶准备四张滤纸。
2)将PDVF膜置于干净的容器中,倒入适量甲醇浸泡1~3分钟,然后浸泡于电转浸泡液中。
3)小心取出两层玻璃之间的胶,切去上层胶。
在电转液中完成如下:电转孔板黑色面铺放海绵一张,依次铺放滤纸两张,胶一块,PVDF膜一张,滤纸再两张,海绵再一张,然后小心扣好孔板,将其黑色面对黑色面扣入电转架,将电转架放入电泳槽。
电泳槽内置小冰盒一个,整个电泳槽置于大冰盒中。
4)接好电源,按蛋白分子量大小调整电压。
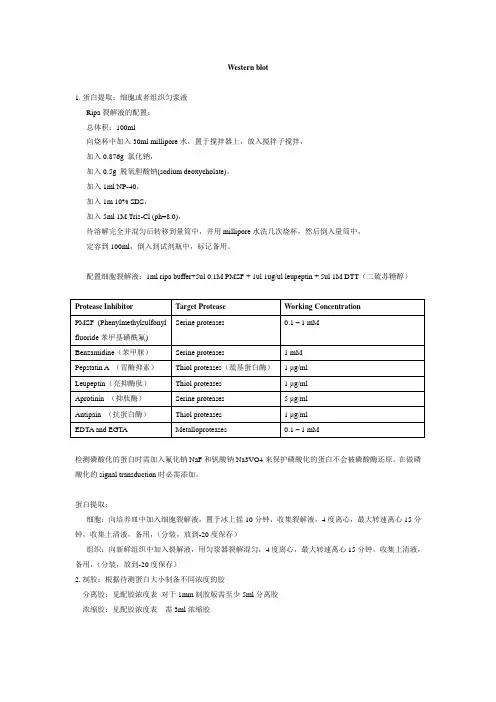
Western blot1.蛋白提取:细胞或者组织匀浆液Ripa裂解液的配置:总体积:100ml向烧杯中加入30ml millipore水,置于搅拌器上,放入搅拌子搅拌,加入0.876g 氯化钠,加入0.5g 脱氧胆酸钠(sodium deoxycholate),加入1ml NP-40,加入1m 10% SDS,加入5ml 1M Tris-Cl (ph=8.0),待溶解完全并混匀后转移到量筒中,并用millipore水洗几次烧杯,然后倒入量筒中,定容到100ml,倒入到试剂瓶中,标记备用。
配置细胞裂解液:1ml ripa buffer+5ul 0.1M PMSF + 1ul 1ug/ul leupeptin + 5ul 1M DTT(二硫苏糖醇)Protease Inhibitor Target Protease Working ConcentrationSerine proteases 0.1 – 1 mMPMSF (Phenylmethylsulfonylfluoride苯甲基磺酰氟)Benzamidine(苯甲脒)Serine proteases 1 mMPepstatin A (胃酶抑素)Thiol proteases(巯基蛋白酶) 1 μg/mlLeupeptin(亮抑酶肽)Thiol proteases 1 μg/mlAprotinin (抑肽酶)Serine proteases 5 μg/mlAntipain (抗蛋白酶)Thiol proteases 1 μg/mlEDTA and EGTA Metalloproteases 0.1 – 1 mM检测磷酸化的蛋白时需加入氟化钠NaF和钒酸钠Na3VO4来保护磷酸化的蛋白不会被磷酸酶还原。
在做磷酸化的signal transduction时必需添加。
蛋白提取:细胞:向培养皿中加入细胞裂解液,置于冰上摇10分钟,收集裂解液,4度离心,最大转速离心15分钟,收集上清液,备用,(分装,放到-20度保存)组织:向新鲜组织中加入裂解液,用匀浆器裂解混匀,4度离心,最大转速离心15分钟,收集上清液,备用,(分装,放到-20度保存)2.制胶:根据待测蛋白大小制备不同浓度的胶分离胶:见配胶浓度表对于1mm制胶版需至少5ml分离胶浓缩胶:见配胶浓度表需3ml浓缩胶H2O 30% acrylamide(丙烯10%APS TEMED 1.5M Tris(8.8) 1.0M Tris(6.8) 酰胺)2.3ml 1.3ml 50ul 5ul 1.3ml8%分离胶(5ml)2.1ml 0.5ml 30ul 3ul 0.38ml5%浓缩胶(3ml)3.玻璃板用酒精棉球擦一遍,晾干,短板向外夹好在绿色架上。

蛋白质印迹法蛋白质印迹法(免疫印迹试验)即Western Blot。
它是分子生物学、生物化学和免疫遗传学中常用的一种实验方法。
其基本原理是通过特异性抗体对凝胶电泳处理过的细胞或生物组织样品进行着色。
通过分析着色的位置和着色深度获得特定蛋白质在所分析的细胞或组织中表达情况的信息。
蛋白免疫印迹(Western Blot )是将电泳分离后的细胞或组织总蛋白质从凝胶转移到固相支持物NC膜或PVDF膜上,然后用特异性抗体检测某特定抗原的一种蛋白质检测技术,现已广泛应用于基因在蛋白水平的表达研究、抗体活性检测和疾病早期诊断等多个方面。
中文名蛋白质印迹法外文名Western Blot蛋白免疫印迹Western Blot类似方法1 Southern Blot 杂交方法类似方法2 Northern Blot 杂交方法使用材料聚丙烯酰氨凝胶电泳⑴原理与Southern Blot 或Northern Blot 杂交方法类似,但Western Blot法采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。
经过PAG(聚丙烯酰胺凝胶电泳)分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。
以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。
该技术也广泛应用于检测蛋白水平的表达。
⑴分类Western Blot 显色的方法主要有以下几种:i. 放射自显影ii. 底物化学发光ECLiii. 底物荧光ECFiv. 底物DAB呈色现常用的有底物化学发光ECL和底物DAB呈色,体同水平和实验条件的是用第一种方法,发表文章通常是用底物化学发光ECL。
只要买现成的试剂盒就行,操作也比较简单,原理如下(二抗用HRP标记):反应底物为过氧化物+鲁米诺,如遇到HRR 即发光,可使胶片曝光,就可洗出条带。

蛋白印记WesternBlotprotocolWestern blot for PAR-2一、试剂的配制1. PMSF(100 mM)的配制:0.1742 g PMSF → 10 ml异丙醇(-20℃保存)2. 裂解液的准备:购自碧云天公司,分强,中,弱三种(三个小瓶,各50 ml).在提取总蛋白之前3-5min,将分装裂解液(强)融化置于冰上,每个肌条300-500ul裂解液不要超过450ul,(1ml加入100 mM的PMSF10 μl(使PMSF 的终浓度为1 mM),置于冰上。
(-20℃保存)3.2X Sample Buffer(要用才配或配好后置于-80℃)1倍DTT加上9倍的2X Laemmli Buffer(1). DTT (Di-dithiothreitol) 1M1.542 g DTT →10 ml的10 mM Sodium acetate(醋酸钠)(pH=5.2)中10 mM Sodium acetate(醋酸钠):0.082 g 无水醋酸钠→ 100 ml dH2O(2). 2X Laemmli Buffer (100 ml) (置于-20°C,经常使用时置于4°C)Glycerol (甘油)20 mlβ-mercaptoethanol (β-me,β-巯基乙醇) 5 ml (恶臭,注意安全)20﹪SDS (十二烷基硫酸钠)10 mlBromophenol Blue(溴酚蓝)20 mg1.5M Tris-Cl(三羟甲基氨基甲烷)pH=6.820 ml (90.855 g Tris→420 ml dH2O,浓盐酸滴定至pH=6.8,定容至500 ml ) dH2O 45 ml4.30% A+B溶液29.2 g Acrilamide (丙烯酰胺)0.8 g Bis-acrilamide (甲叉双丙烯酰胺)Add dH2O dilute to 100 mL and filter 避光4°C储存注意:该两种药品有毒,配药时注意安全PS. Acrylamide 及Bisacrylamide 是neurotoxin会穿过皮肤,配药时要穿实验衣及戴口罩。

westernblot原理及过程
1、Western Blot的原理是:蛋白质是带电的,在聚丙烯酰胺凝胶中通过SDS-PAGE分离蛋白质后,蛋白质被固定在凝胶中。
当凝胶被转移到支持物(例如NC膜或PVDF膜)上时,蛋白质在膜上保留其电荷。
通过针对特定氨基酸序列的特异性抗体作为探针检测蛋白质的位置。
这种技术的作用是对细胞或组织提取的蛋白混合物中的某一特异蛋白进行鉴别和半定量分析。
2、Western Blot的过程大致分为以下几步:
蛋白提取:从细胞或组织中提取总蛋白质混合物。
蛋白定量:确定凝胶中蛋白质的浓度。
SDS-PAGE电泳:通过SDS-PAGE分离蛋白质混合物。
电转印:将分离的蛋白质从凝胶转移到支持物(例如NC膜或PVDF 膜)上。
封闭:用含有去污剂和牛血清白蛋白的溶液处理膜,以封闭膜上的任何未结合位点。
抗原-抗体免疫反应:使用特异性抗体检测目标蛋白质的位置。
蛋白检测:使用化学发光剂检测膜上的目标蛋白质。

一.绘制BSA标准曲线(目的:测蛋白浓度)1.方法:BCA法(生工)2.需要配的试剂:1× PBS溶液10× PBS溶液:Na2HPO48 mMNaCl 136mMKH2PO4 2mMKCl 2.6mM3.注意:① 用分光光度计测定A562吸光值时,所需样品最小体积为1ml,即,加样体积为:1× PBS—0.5ml,BCA工作液—0.5ml;① 由于缓冲液1× PBS与BCA工作液是1:1等体积加的,所以,BSA被稀释为初次用1× PBS配制的梯度浓度的1/ 2,即,在绘制标准曲线时,BSA的终浓度需要÷ 2,切记!二.提取总蛋白1.试剂:配制时佩戴手套、口罩。
(1)单一母液:① 1M Tris (pH=8.0):4℃保存称取12.114g Tris-base,加少于100ml蒸馏水溶解,调pH后,定容至100ml。
① 0.5M EDTA (pH=8.0):4℃保存称取14.61g EDTA,加少于100ml蒸馏水,加固体NaOH至pH约为8.0时,EDTA方开始溶解,溶液状态变化过程:白色乳浊液——白色胶状物——白色乳浊液——无色透明液。
① 20% SDS:常温保存称取20g SDS,加蒸馏水定容至100ml.④100× PMSF (100 mM): 4℃保存称取261.3 mg PMSF,加15 ml 异丙醇溶解。
(2)复合母液:2× extraction buffer : 40ml1M Tris (pH=8.0) 2 ml0.5 M EDTA (pH=8.0) 160 μl20% SDS 4 mlWater 33.84 ml2.取样:取细胞浓度为2×107的藻液1ml于1.5ml EP管中,厌氧箱中离心1-2min,将沉淀物于液氮中速冻后置于-80①保存;3.提取总蛋白:3.1 根据样品数量计算所需的提取试剂体积:1个样品——1ml 1× extraction buffer3.2 配制1× extraction buffer:试剂:① 2× extraction buffer① 100× PMSF3.3 提取:将-80①保存的样品取出,置于冰上,加入步骤2.2配制的1× extraction buffer 1ml, 涡旋至EP管底部无沉淀黏着后,4①,12,000 rpm离心10 min,用1ml移液枪小心吸取上清于另一干净的1.5 ml EP管中,置于冰上。
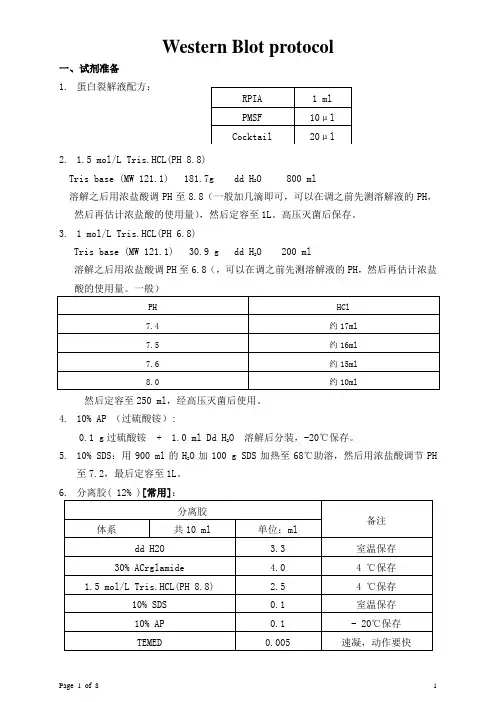
Western Blot protocol一、试剂准备1.蛋白裂解液配方:2. 1.5 mol/L Tris.HCL(PH 8.8)O 800 mlTris base (MW 121.1) 181.7g dd H2溶解之后用浓盐酸调PH至8.8(一般加几滴即可,可以在调之前先测溶解液的PH,然后再估计浓盐酸的使用量),然后定容至1L。
高压灭菌后保存。
3. 1 mol/L Tris.HCL(PH 6.8)Tris base (MW 121.1) 30.9 g dd HO 200 ml2溶解之后用浓盐酸调PH至6.8(,可以在调之前先测溶解液的PH,然后再估计浓盐酸的使用量。
一般)然后定容至250 ml,经高压灭菌后使用。
4.10% AP (过硫酸铵):O 溶解后分装,-20℃保存。
0.1 g过硫酸铵 + 1.0 ml Dd H2O加100 g SDS加热至68℃助溶,然后用浓盐酸调节PH 5.10% SDS:用900 ml的H2至7.2,最后定容至1L。
6.分离胶( 12% )[常用]:7.浓缩胶( 5% ):8. 5 × Running Buffer(储存液):9. 1 × Running Buffer (工作液):200 ml的5 × Running Buffer + 800 ml的ddO。
H210.转膜缓冲液:3.03 g Tris base + 14.4g甘氨酸溶解在少量的蒸馏水中,完全溶解后加100ml的甲醇,再用蒸馏水定容至1L。
11.10 × TBS ( 储存液) :24.2 g Tris base + 80 g NaCl 溶解,用Hcl调PH至7.6,最后定容至1L。
O。
12.1 × TBS (工作液):100 ml的10 × TBS + 900 ml的dd H213.TBST:含0.1% 吐温—20的1 × TBS。

WesternBlot最全攻略:从protocol到问题解决蛋⽩免疫印迹(western blotting)⼀般由凝胶电泳、样品的印迹和免疫学检测3个部分组成,是根据抗原抗体特异性结合检测样本中某种蛋⽩的⽅法,与ELISA不同的地⽅在于WB是⼀种半定量的检测⽅法。
操作流程如下:下⾯详细介绍⼀下每个操作步骤:⼀、样本的制备(1)样本⼀般是组织和细胞,根据样本类型的不同,选择合适的裂解液:样本类型裂解液全细胞NP-40、RIPA细胞质(可溶性)Tris-HCl细胞质(细胞⾻架)Tris-Triton膜结合部分NP-40、RIPA细胞核RIPA、核裂解液线粒体RIPA(2)抑制剂,根据研究的⽬的蛋⽩进⾏选择,假如要检测磷酸化的蛋⽩含量,就要对磷酸酶进⾏抑制,现在很多公司都有多种抑制剂的混合物抑制剂靶点浓度2µg/ml抑肽酶胰蛋⽩酶、⾎纤维蛋⽩溶酶亮抑酶肽溶酶体1-10µg/ml胃蛋⽩酶抑制剂A Asp蛋⽩酶1µg/mlPMSF Asp蛋⽩酶1mMEDTA镁和锰⾦属蛋⽩酶1-5mMEGTA钙⾦属蛋⽩酶1mM氟化钠丝氨酸和苏氨酸磷酸酶5-10mM原钒酸盐酪氨酸磷酸酶1mM焦磷酸盐丝氨酸和苏氨酸磷酸酶1-2mMβ-⽢油磷酸盐丝氨酸和苏氨酸磷酸酶1-2mM(3)要加⼊loading buffer(BME、DTT,⽬的是减少蛋⽩⾼级结构的形成)主要成分主要功能SDS使蛋⽩变性并带上负电荷BME或DTT减少⼆硫键的形成溴酚蓝离⼦型染料,可观察蛋⽩迁移⽢油增加样品密度,起沉降作⽤(4)煮沸变性:⼀般是99°C,10min,⽔浴锅煮沸时,防⽌EP管盖弹开,使⽔浴锅内的⽔进⼊。
⼆、凝胶电泳(1)配胶。
这⾥说的都是SDS变性胶,先配分离胶,再配浓缩胶。
根据⽬标蛋⽩分⼦量的⼤⼩,选择合适的分离胶浓度,分⼦量越⾼,分离胶的浓度越低。
浓缩胶⼀般⽤的都是5%。
配胶时需注意以下⼏点:①玻璃板清洗后,为了使表⾯的⽔快速晾⼲,可以在烘箱中放置⼏分钟,拿出来的时候⼀定要晾到室温再灌胶,不然很容易产⽣⽓泡;②配胶⽤的试剂⼀般都是在4度冰箱保存,当我们按⽐例配好混合液的时候,要等混合液恢复室温时,再灌胶,不然也很容易产⽣⽓泡;③加⽔液封时,速度要慢,可⽤1ml枪沿着胶板上沿反复移动,不然很容易将分离胶冲变型。

免疫印迹(Western Blot)操作规程实验目的:免疫印迹(Western Blot)是将蛋白质转移到膜上,然后利用抗体进行检测。
对已知表达蛋白,可用相应抗体作为一抗进行检测,对新基因的表达15.14g Tris 10 ml 10% SDS 溶解后调节 pH 到6.8 需要注意的是要在低温调节pH电极缓冲液:1L 6g Tris 28.8g Gly 溶解后加10ml 10% SDS 定容到1L样品处理液:须配制20*buffer 200mM Tris 20mM EDTA pH 8.05*loading buffer(see Takara)组分:250mM Tris-HCL(PH6.8) 10%SDS 0.5%BPB 50%甘油 5%β-巯基乙醇(2-ME)1M Tris-HCL(PH6.8) 1.25mlSDS 0.5gBPB 25mg甘油 2.5ml去离子水溶解后定容到5ml小份(500ul/份)分装后,于室温保存。
使用前将25ul的2-ME加到每小份中,加入2-ME的Loading buffer可在室温下保存一个月左右。
实验操作程序:样品处理由于蛋白酶抑制剂可影响蛋白定量,且新鲜蛋白很少降解,故可不加,如加按建议比例即可。
提取磷酸化的蛋白还需加Na3VO4 0.1mM(sigma)及NaF25mM(sigma))。
注意SDS终浓度勿超过10%。
对于心脏,肌肉等碎屑较多的组织可用5%的SDS,肝肾等组织2%即可。
蛋白裂解液:PBS+1%Triton+1%SDS+1*Protease inhibitor cockerIII (蛋白酶抑制剂,MEK:59134-1set)+Na3VO4 0.1mM(sigma)及NaF 25mM(sigma))。
或者Phosphatase Inhibitor Cocktail III (磷酸酶抑制剂,biovision:K276-1EA) 培养的细胞(定性):1. 去培养液后用温的PBS冲洗2~3遍(冷的PBS有可能使细胞脱落)。


western blot实验内容Western Blot实验内容可以简单描述为一种生物化学分析技术,主要用于检测特定蛋白质在细胞或组织中的存在和表达水平。
该实验通常包括以下步骤:1. 样品制备:首先,从细胞培养物或组织中提取蛋白质。
这可以通过细胞裂解和蛋白质提取试剂盒等方法来完成。
2. SDS-PAGE电泳:将提取的蛋白质样品通过SDS-PAGE凝胶电泳进行分离。
在SDS-PAGE中,样品中的蛋白质按照大小被分离成不同的带状条带。
3. 转印:将分离的蛋白质通过电泳转印技术转移到聚丙烯酰胺薄膜(或称为膜)上。
膜可以是尼龙膜或聚乙烯膜。
4. 阻断:将膜置于含有蛋白质阻断剂(如牛血清蛋白或脱脂奶粉)的缓冲液中,以阻止非特异性结合。
5. 一抗孵育:将膜与特异性抗体(一抗)一起在某种缓冲液中孵育。
这些一抗可以结合到目标蛋白质上。
6. 洗涤:通过多次洗涤,去除与一抗非特异性结合的蛋白质。
7. 二抗孵育:膜与与一抗的物种不同的次级抗体(二抗)一起在缓冲液中孵育。
二抗与一抗结合,形成复合物。
8. 洗涤:去除与二抗非特异性结合的蛋白质。
9. 显色:将特定酶底物添加到膜上,使目标蛋白质变色。
例如,常用的酶底物是辣根过氧化物酶(HRP)和4-氨基乙基硅烷(DAB),可以生成棕色的色斑。
10. 图像获取和分析:使用相应的设备(如基于膜的成像系统)捕捉膜的图像,并使用分析软件定量分析蛋白质的表达水平。
通过Western Blot实验,研究人员能够检测和定量不同细胞组分中蛋白质的存在和表达水平,从而深入了解生物系统的功能和调控机制。
Western Blot protocolSDS-PAGE:SDS-PolyAcrylamide Gel Electrophoresis【原理】Western Blot又称免疫印迹,是指将蛋白样品转移到固相载体上,而后利用相应的抗体来检测目的蛋白的一种方法。
是检测混合样品中单一特定蛋白的常用技术。
Western Blot间接法基本原理:首先利用SDS-PAGE对蛋白质样品进行分离,然后转移到固相载体(例如PVDF膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。
转移后的PVDF膜就称为一个印迹(blot),用于对蛋白质的进一步检测。
印迹首先用蛋白溶液(如5%的BSA 或脱脂奶粉溶液)处理以封闭PVDF膜上剩余的疏水结合位点,而后用所要研究的蛋白质的抗体(一抗)处理,印迹中只有待研究的蛋白质与一抗特异结合形成抗原抗体复合物,而其它的蛋白质不能与一抗结合,这样清洗除去未结合的一抗后,印迹中只有待研究的蛋白质的位置上结合着一抗。
处理过的印迹进一步用适当标记的二抗处理,二抗是指一抗的抗体,如一抗是从鼠中获得的,则二抗就是抗鼠IgG的抗体。
处理后,带有标记的二抗与一抗结合形成抗体复合物可以指示一抗的位置,即是待研究的蛋白质的位置。
目前有结合各种标记物的抗特定IgG 的抗体可以直接购买作为二抗,最常用的一种是酶连的二抗,印迹用酶连二抗处理后,再用适当的底物溶液处理,当酶催化底物生成有颜色的产物时,就会产生可见的区带,指示所要研究的蛋白质位置。
该技术广泛应用于检测蛋白水平的表达。
在Western Blot实验中,还有另一种方法,就是直接标记一抗,再用底物显色,这种方法叫直接法。
与用二抗的间接法相比有诸多不足,标记二抗可用于很多种不同特异性的一抗,避免了标记很多一抗的需要,同时因为一抗结合不止一个二抗分子,所以二抗可以增强信号。
所以一般情况下都釆用间接法进行检测。
【实验耗材】•PVDF膜[MILLIPORE (IPVH00010)]•NC膜•滤纸[BIO-RAD (1703967)]【实验仪器】•PowerPac Basic电泳仪[BIO-RAD (041BR89156)]•Trans-Blot Turbo System 半干转转膜仪[BIO-RAD (690BR006827)]自制胶通常使用Bio-rad半干转转膜仪•iBlot Dry Blotting System 干转转膜仪[Invitrogen (25-0912)]需用Invitrogen 预制转印包•Chemi Doc XRS+显影仪[BIO-RAD (721BR04128)]【试剂】••5×量取100mL of 5×电泳液至500mL量筒中,加超纯水至500mL刻度线,配好后的电泳液在1个月内使用。
WesternBlot原理、protocol一、组织、细胞总蛋白抽提:参考相关裂解液;二、蛋白浓度测定:BCA法,这个结果要参考说明书!准备:BCA试剂,BSA,PBS/生理盐水,96孔板,100ul、20ul、10ul移液*及*头,1.5ml及4ml EP管,冰河1、配制BSA蛋白标准:配0.5mg/ml BSA,将25mg/ml的母液用RIPA 稀释成0.5mg/ml(RIPA可以换成PBS,下面的也是);2、将BCA试剂A与B按50:1(2450 A+49ul B)充分混匀,配置成适量BCA工作液(4℃);3、稀释BSA蛋白标准品和待测蛋白样品:用PBS稀释至20ul,再向每孔加入200ul BCA(在20-1000μg/ml浓度范围内有较好的线性关系)注:按RIPA-BSA-样本-BCA的顺序加入,操作要迅速!4、锡纸遮盖,37℃恒温箱(或酶标仪)放置30min;5、置于酶标仪上测定A562(或A570),用excel建立标准曲线然后计算出蛋白浓度;注意:数据分析可以在Excel里做也可以在GraphPad Prism软件里做,R平方值越接近1数据越好三、Western blot步骤:(一)、玻板的清洗:自来水→75%酒精擦拭→ddH2O→竖放晾干/吹干(做胶的一面朝内)【勿用手指触摸玻板内侧面】装板:低的一面向内,确认玻板底部平齐,插入斜楔板压实卡紧,加ddH2O不漏,滤纸吸干水;(二)、配胶( 1.0mm):分离胶 5% 浓缩胶, 3 ml/块胶注意:①、用*吸取胶,沿玻璃板一侧注入,先快后慢,*头不要打到底以防产生气泡,胶面升到绿色板上缘,用75%酒精封闭液面;②、室温放置约30min使胶充分凝固,待水和胶面之间有肉眼可见的折线,小心倒掉上层水,并用滤纸小心吸干水,勿碰触到胶面;③、TEMED作用为促凝胶,如其他试剂放置时间较长或气温较低不易凝胶时,可适量多加1~2倍 TEMED,或者多加点AP,胶就会很快凝固的;④、颠倒混匀后灌胶(约2ml),插入梳子,室温放置约30 min 后,将玻板取出装入电泳装置,竖直向上将梳子拔出,放入电泳槽润湿板子,再拿出将玻璃板卡准;补充下浓缩胶的作用:链接:/content/12/0319/10/2867540_1955684 87.shtml(三)、点样及电泳:1、用10 ul *吸取电泳液,倾斜45°反复吹打上样孔,去掉胶的残渣;2、按顺序加样本,侧边孔加5ul marker,多余孔加入2ul loading buffer,迅速上样避免弥散;上样的量可以根据所测蛋白浓度来调整,有的建议20-40ug,有的50-100ug,其实个人觉得40-80ug比较适合的,主要看你样品浓度吧!3、恒压80V 约20min;待样品进入二胶的分界线时(时间也不一定20min,跑到交界处就可以改变电压),120V 约90min(设I=200,T=2:00)(100mA的条件也跑过-六一产的);4、溴酚蓝跑至玻板底部,且Marker条带分的足够开时,停止电泳;(四)、转膜:准备(提前30min):1×转膜液、培养皿、甲醇、PVDF膜、剪刀镊子、尺子、切胶板、托盘、转膜夹、冰袋及冰。
蛋白质印迹/Western blotting实验操作步骤一、总蛋白的提取单层贴壁细胞总蛋白的提取:1)吸除培养液2)每皿细胞加4℃预冷的 PBS。
平放轻轻摇动 1min 洗涤细胞,然后弃去洗液。
重复上操作两次,共洗细胞三次以洗去培养液。
将PBS弃净后把培养瓶置于冰上。
(PBS会降低细胞裂解液的效价和总蛋白的浓度)3)加裂解液于冰上裂解 30 min,为使细胞充分裂解,培养瓶要经常来回摇动(可放置在4℃摇床裂解)。
4)裂解完后,用干净的刮棒将细胞刮于培养瓶的一侧(动作要快),然后用枪将细胞碎片和裂解液移至 1.5mL 离心管中。
(整个操作尽量在冰上进行)5)在EP管中将细胞震碎(10s/次,3次)6)于4℃下 12000rpm 离心 20-30 min。
(离心机提前预冷至4℃)7)将离心后的上清分装转移倒 1.5mL 的离心管中放于-20℃保存。
二、BCA法测蛋白浓度1)将BCA protein assay每孔 A液200μL,B液4μL混合,96孔板每孔加入22.5μLdd水,2.5μL蛋白提取液,200μLA+B混合液2)在烘箱中37℃,90r,孵育30min3)使用酶标仪测出吸光度后,使用公式y=0.9154x-0.118计算出蛋白浓度(浓度需要×10)4)将蛋白配成等浓度等体积(使用配置好的裂解液配),按照4:1加入5X loading buffer然后煮5min(100℃),放入-20℃保存三、SDS-PAGE电泳板子1.5mm,梳子1.5mm1)清洗玻璃板:一只手扣紧玻璃板,另一只手蘸点洗衣粉轻轻擦洗。
两面都擦洗过后用自来水冲洗2)验漏:玻璃板对齐后放入夹中卡紧,然后垂直卡在架子上,加满水验漏3)灌胶:验漏结束后用纸吸干水分,按方法配制下层胶(4mL+4mL+80μLAP),灌胶时,可用 1mL 枪吸取胶沿玻璃放出,待胶面升到绿带中间线高度时即可。
然后胶上加1 mL水,液封后的胶凝的更快。
蛋⽩质免疫印迹(WesternBlot)实验步骤和原理及注意事项蛋⽩质免疫印迹(Western Blot )实验步骤和原理及注意事项1.收集蛋⽩样品(Protein sample preparation)可以使⽤适当的裂解液。
收集完蛋⽩样品后,为确保每个蛋⽩样品的上样量⼀致,需要测定每个蛋⽩样品的蛋⽩浓度。
根据所使⽤的裂解液的不同,需要采⽤适当的蛋⽩浓度测定⽅法。
因为不同的蛋⽩浓度测定⽅法对于⼀些去垢剂和还原剂等的兼容性差别很⼤。
BCA法。
2. 电泳(Electrophoresis)(1) SDS-PAGE凝胶配制(2) 样品处理在收集的蛋⽩样品中加⼊适量浓缩的SDS-PAGE蛋⽩上样缓冲液。
例如2X或5X的SDS-PAGE蛋⽩上样缓冲液。
使⽤5X的SDS-PAGE蛋⽩上样缓冲液可以减⼩上样体积,在相同体积的上样孔内可以上样更多的蛋⽩样品。
100℃或沸⽔浴加热3-5分钟,以充分变性蛋⽩(根据蛋⽩分⼦的⼤⼩,煮沸时间可适当变化,⼀般不低于5min。
煮沸只是变性蛋⽩,⽽不是分解,⼀般加了抑制酶不会分解。
煮沸对于SDS-PAGE凝胶电泳是必须的,只有煮沸,才能消除蛋⽩质的⽴体⼆级结构,伸展为⼀维线性结构,所以⼀般来讲⼆聚体都会解体,才能完全按照分⼦量跑电泳,加的蛋⽩Marker才有指⽰分⼦量的意义。
蛋⽩样品变性后与SDS充分结合,SDS使每个氨基酸带相同的电荷,使整个蛋⽩呈线性结构. 抗体因为要是线性表位结合的,100度煮10min 后13000,离⼼5分钟,取上清电泳,因为沉淀会导致拖尾.也可以取上清到另⼀管,4度可以放⼀周备再次电泳)。
(3)电泳i.清洗玻璃板:⼀只⼿扣紧玻璃板,另⼀只⼿蘸点洗⾐粉轻轻擦洗。
两⾯都擦洗过后⽤⾃来⽔冲,再⽤蒸馏⽔冲洗⼲净后⽴在筐⾥晾⼲。
ii.灌胶与上样(1)玻璃板对齐后放⼊夹中卡紧。
然后垂直卡在架⼦上准备灌胶。
(操作时要使两玻璃对齐,以免漏胶。
)(2)配10%分离胶,加⼊TEMED后⽴即摇匀即可灌胶。
Western blot实验方案【实验目的】了解western blot原理,掌握有关的操作方法。
【实验原理】Western Blot法采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。
经过PAGE(聚丙烯酰胺凝胶电泳)分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。
以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。
【实验仪器、材料和试剂】(1)仪器、用具:高速冷冻离心机、振荡器、-80℃冰箱、-20℃冰箱、酶标仪、水浴锅、涡旋器、SDS电泳仪、PH自动控制加液机、摇床、半干转膜仪、湿转膜仪、凝胶成像系统、托盘、量筒、1L烧杯、小烧杯、冰盒、孵育盒、纤维垫、玻璃棒、有齿镊子、无齿镊子、表面皿、搪瓷盘、移液器、细胞刮刀、计时器(2)耗材:15mL离心管、50mL离心管、1.5mL EP管、96孔板、普通滤纸、whatman 3mm 滤纸、PVDF膜、一次性枪头、棉球、保鲜膜(3)材料:人乳腺癌MCF-7细胞株、人乳腺癌MDA-MB-231细胞株(4)试剂:75%酒精、0.25%胰蛋白酶、PBS、RIPA裂解液、PMSF、bradford蛋白定量检测试剂盒、1.0mol/L Tris•HCl (pH6.8)、1.5mol/L Tris•HCl(pH8.8)、10%AP、TEMED原液、30%丙烯酰胺(29:1)、10%SDS阴离子去污剂、Sample Loading Buffer (5X)、BSA牛血清白蛋白、Running Buffer(5X)、transfer buffer、无水甲醇溶液、脱脂奶粉、10XTBS缓冲液、Tween-20、β-actin、一抗、带HRP二抗、DAB显色试剂盒、Stripping Buffer膜再生利用液(5)需要配制:分离胶、浓缩胶、封闭液(抗体稀释液)、洗脱液需要稀释:加样缓冲液、电泳缓冲液、转膜液其余按照说明书操作。
Western Blot
Solutions:
Solution 1 100ml
Acrylamide 29.2g
Bisacrylamide 0.8g
Add ddH2O to 100ml. Use filter paper to filter. Store at 4℃
Solution 2 200ml
1.5 M Tris pH 8.8 (measure Tris 36.3g) Add ddH2O to 200 ml. Autoclave.
Solution 3 100ml
0.5 M Tris pH6.8 (measure Tris 6g) Add ddH2O to 100 ml. Autoclave.
Solution 4
10%SDS (measure SDS 10g) Add ddH2O to 100 ml. Autoclave.
For gel making
10% separation Gel 5ml
Solution 1 1.667ml
Solution 2 1.25ml
Solution 4 0.05ml
ddH2O 2.017ml
10% AP 50μl
TEMED 5μl
4% stacking Gel 2ml
Solution 1 0.266ml
Solution 3 0.5ml
Solution 4 0.02ml
ddH2O 1.22ml
10% AP 15μl (AP: Ammonium persulfate, measure 0.1g to 1ml ddH2O) TEMED 1.5μl
10* running buffer 1000ml
Tris 30g
Glycine 144g
10%SDS 100ml or measure SDS 10g
Add ddH2O to 1000ml
For 1* running buffer, Add 100ml 10*buffer to 900ml ddH2O
10* Transfer buffer 1000ml
Tris 30g
Glycine 144g
Add ddH2O to 1000ml
For 1* Transfer buffer,
10* stock 100ml
Methanol 200ml
ddH2O 700ml
Stripping buffer 1000ml
0.2 M glycine (measure 15g glycine)
Use HCl to adjust pH to 2.5 (nearly 9.5ml HCl)
Store at 4℃
4*sample buffer w/DTT 50ml
Glycerol 25.2g (or 20ml)
bromophenol blue 0.02g ( you can add less than 0.02g,0.01g maybe enough) SDS 4g
1M Tris,pH6.8 20ml
DTT powder 3.1g
ddH2O up to 50 ml
Store at -20℃.
Western blot
1.Protein extraction
1 leaf, 20- 30μl 4*sample buffer, grounding,
90℃-100℃,boil for 5min. put on ice, then high speed centrifuge for 10-15min.
2.SDS-PAGE 86v for stacking gel, 136v for separating gel
3.Western.
a. Put your nylon membrane and gel in 1*transfer buffer for 5min, then you can make
the sandwich. From button to up:
Black color stands for cathode, then put filter paper (if it is thick one, use one layer; if it is thin one, use two layers), then your gel, then your nylon membrane, then filter paper, then Red color which stands for anode. All progress should be done in the transfer buffer, and need to remove bubbles. 86v for 50 min, or 8v for overnight. (for wet),if Semi-Dry, use 20v for 50min.
(Use Ponceau stain to check transfer.)
b. 5% milk/PBST block for 1h-2h.
c. Wash with PBST 1-2 times, 10sec
d. Incubate in the first Antibody for 1h-2h
The first antibody is diluted in PBST or 1%milk/PBST.
Myc 1:4000 GFP1:3000-1:5000 CRY1 1:3000-1:5000 CRY2 1:3000
e. Wash with PBST for 10 min,3 times or 6 min,3 times
After you remove your first antibody (usually we put this antibody/PBST in 4℃ for one week, in this week, you can reuse for twice, don’t use it more than 3times),use PBST to wash for secs and then start the wash step. For myc, I usually wash 2times,each time no more than 5mins.
f. Incubate in second antibody for 1h. For myc, use anti-monse,1:4000; for
cry1/cry2/GFP/4E/H1, use anti-rabbite,1:10000.
g. Wash with PBST for 10 min,3 times or 6 min,3 times.
h. Develop film. 3ml solution A + 3ml solution B + 1.8μl H2O2 (this one should
add fresh before you use) put your nylon membrane in this solution for 1min , then use preservative film to wrap the membrane and put into exposure holder, and put a film on it, exposure different time as you wish. then put the film into the develop machine.
I got this method from Hongtao, it is very useful.
Lixu
10xPBS 1 L
NaCl 80g
KH2PO4 2g
KCl 2g
Na2HPO4.12H2O 28.3g Na2HPO4 11.4g
ddH2O
Ph 7.4
1xPBST 1L
10xPBS 100ml
Tween 1ml
ddH2O
丽春红染料100ml
三氯乙酸3g
丽春红0.2g
ddH2O
考马斯亮蓝1L
G250 1g
甲醇500ml 醋酸100ml ddH2O
考马斯亮蓝洗脱液1L 甲醇250ml
醋酸100ml
ddH2O。