骨科手术器械技术审评规范修订-2012版
- 格式:doc
- 大小:1.99 MB
- 文档页数:8

骨科手术器械(二类)产品技术审评规范(2017版)附件3骨科手术器械(第二类)产品注册审评规范(2017版)本审评规范旨在指导和规范骨科手术器械(第二类)产品的技术审评工作,帮助审评人员理解和掌握该类产品原理/机理、结构、性能、预期用途等内容,把握技术审评工作基本要求和尺度,对产品安全性、有效性作出系统评价。
本审评规范所确定的核心内容是在目前的科技认识水平和现有产品技术基础上形成的。
因此,审评人员应注意其适宜性,密切关注适用标准及相关技术的最新进展,考虑产品的更新和变化。
本审评规范不作为法规强制执行,不包括行政审批要求。
但是,审评人员需密切关注相关法规的变化,以确认申报产品是否符合法规要求。
一、适用范围本审评规范适用于第二类骨科手术器械,一般包括:使用过程中与椎间隙直接接触的骨科手术器械;骨科内窥镜用手术器械;以无菌形式提供的骨科手术器械;由网电源、电池或气源提供所需机械动力的骨科手术刀具(如钻、铣、锯、磨、刨等);第二类骨科手术器械包等。
本审评规范不适用于骨科外固定器械和一次性使用骨科手术器械,一次性使用骨科手术器械可参考本审评规范的相关内容。
骨科手术器械产品类代号为6810。
二、技术审查要点(一)产品名称的要求骨科手术器械产品名称依据《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)确定。
骨科手术器械的名称可按作用对象和预期用途等方式来命名,如椎管铲刀、颈椎刮匙、椎板咬骨钳、无菌骨牵引针。
由网电源、电池或气源提供所需机械动力的骨科手术器械,可命名结构为:电动/电池式+刀具对象+刀具名称,如电动胸骨锯、电动骨钻。
骨科手术器械包类产品以体现产品组成、功能用途为基本原则,由多种类别器械组成,一般应以其主要预期用途来命名,如椎间融合手术器械包。
(二)产品的结构组成骨科手术器械产品应明确产品结构组成,宜采用图示方式表述产品各组成部分,并明确所用材料。
由网电源、电池或气源提供所需机械动力的骨科手术器械一般由单一或多件与手机配合使用的刀具组成(如:钻类、铣类、磨类、锉类、锯类、刨类)。

人工膝关节置换技术管理规范(2012年版)为规范人工膝关节置换技术的临床应用,保证医疗质量和医疗安全,根据《医疗技术临床应用管理办法》,制定本规范。
本规范为医疗机构及其医师开展人工膝关节置换技术的基本要求。
本规范所称人工膝关节置换技术包括全膝关节置换及部分膝关节置换技术,不包括膝部肿瘤切除后的假体重建技术。
一、医疗机构基本要求(一)医疗机构开展人工膝关节置换技术应当与其功能、任务相适应。
(二)三级医院,有卫生行政部门核准登记的骨科诊疗科目及其他相关科室和设备。
1.骨科。
(1)开展骨科临床诊疗工作10年以上,床位不少于50张,设有关节外科专科病房或专业组,关节外科床位不少于20张。
(2)每年完成人工膝关节置换手术50例以上。
2.开展人工膝关节置换手术的手术室。
(1)有至少1间手术室达到I级洁净手术室标准(手术。
级)1000级层流、周边区100区(2)手术室使用面积30平方米以上,布局合理。
(3)配有经国家食品药品监督管理局批准的满足人工膝关节置换手术需要的手术器械。
(4)配备符合放射防护条件的C臂X线机。
3.其他相关科室和设备。
(1)设有麻醉科、重症监护室、心血管内科、呼吸内科、内分泌科及康复科等专业科室或专业医师,具备全身合并症、并发症的综合处理和抢救能力。
(2)具备CT、床边X线摄影机、术后功能康复系统。
(三)具有专业骨科医师队伍,其中包括至少2名副主任医师以上专业技术职务任职资格的医师,人员梯队结构合理。
(四)拟开展人工膝关节置换技术的新建或者新设骨科的三级医院,应当符合本规范的人员、科室、设备、设施条件,并向省级卫生行政部门提出申请,通过省级卫生行政部门组织的临床应用能力评估后方可开展。
二、人员基本要求(一)开展人工膝关节置换技术的医师。
1.取得《医师执业证书》,执业范围为外科专业。
2.有10年以上骨科临床诊疗工作经验,具有副主任医师以上专业技术职务任职资格。
3.近3年每年参与完成膝关节置换手术至少20例。
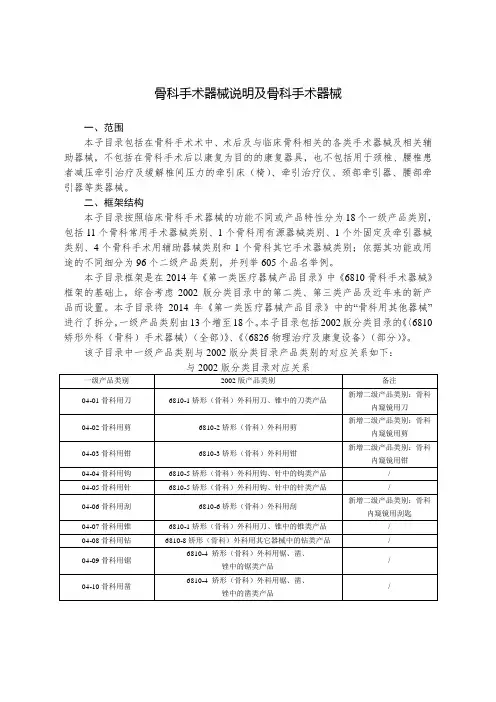
骨科手术器械说明及骨科手术器械一、范围本子目录包括在骨科手术术中、术后及与临床骨科相关的各类手术器械及相关辅助器械,不包括在骨科手术后以康复为目的的康复器具,也不包括用于颈椎、腰椎患者减压牵引治疗及缓解椎间压力的牵引床(椅)、牵引治疗仪、颈部牵引器、腰部牵引器等类器械。
二、框架结构本子目录按照临床骨科手术器械的功能不同或产品特性分为18个一级产品类别,包括11个骨科常用手术器械类别、1个骨科用有源器械类别、1个外固定及牵引器械类别、4个骨科手术用辅助器械类别和1个骨科其它手术器械类别;依据其功能或用途的不同细分为96个二级产品类别,并列举605个品名举例。
本子目录框架是在2014年《第一类医疗器械产品目录》中《6810骨科手术器械》框架的基础上,综合考虑2002版分类目录中的第二类、第三类产品及近年来的新产品而设置。
本子目录将2014年《第一类医疗器械产品目录》中的“骨科用其他器械”进行了拆分,一级产品类别由13个增至18个。
本子目录包括2002版分类目录的《〈6810矫形外科(骨科)手术器械〉(全部)》、《〈6826物理治疗及康复设备〉(部分)》。
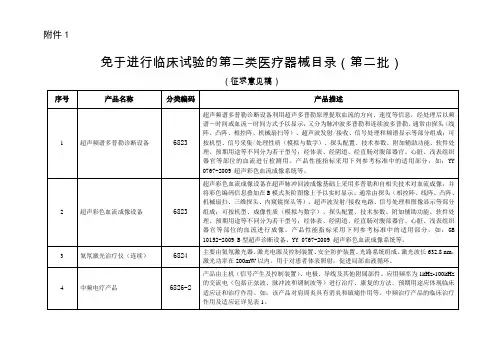
该子目录中一级产品类别与2002版分类目录产品类别的对应关系如下:三、其他说明(一)骨髓抽吸和活检系统电钻,规范为第二类管理。
(二)与有源设备(如电动骨钻、电动骨锯、气动骨钻)连接使用的钻头、刀头、锯片、扩髓器、刨刀、磨头等配套工具,规范为第一类管理。
(三)髌骨爪,尖端侵入人体,且滞留时间大于30天,规范为第三类管理。
(四)接触椎间隙的非无菌提供的骨科重复使用手术器械,管理类别由第二类降为第一类。
(五)开口用锥(如手锥),管理类别由第二类降为第一类。
(六)对于在手术操作过程中本子目录可能接触中枢神经系统的非无菌提供的骨科用凿、骨科用锉、骨科用铲、脊柱外科定位/导向/测量器械、脊柱外科开孔扩孔器械、脊柱外科神经根探子、脊柱外科植骨块嵌入器、脊柱外科椎弓根钉尾部切断器、脊柱手术通道器械、脊柱外科椎体复位器、骨科剥离保护器、骨科组织保护器具,管理类别由第二类降为第一类。

附件3骨科手术器械(第二类)产品注册审评规范(2017版)本审评规范旨在指导和规范骨科手术器械(第二类)产品的技术审评工作,帮助审评人员理解和掌握该类产品原理/机理、结构、性能、预期用途等内容,把握技术审评工作基本要求和尺度,对产品安全性、有效性作出系统评价.本审评规范所确定的核心内容是在目前的科技认识水平和现有产品技术基础上形成的.因此,审评人员应注意其适宜性,密切关注适用标准及相关技术的最新进展,考虑产品的更新和变化.本审评规范不作为法规强制执行,不包括行政审批要求.但是,审评人员需密切关注相关法规的变化,以确认申报产品是否符合法规要求.一、适用范围本审评规范适用于第二类骨科手术器械,一般包括:使用过程中与椎间隙直接接触的骨科手术器械;骨科内窥镜用手术器械;以无菌形式提供的骨科手术器械;由网电源、电池或气源提供所需机械动力的骨科手术刀具(如钻、铣、锯、磨、刨等);第二类骨科手术器械包等.本审评规范不适用于骨科外固定器械和一次性使用骨科手术器械,一次性使用骨科手术器械可参考本审评规范的相关内容.骨科手术器械产品类代号为6810.二、技术审查要点(一)产品名称的要求骨科手术器械产品名称依据医疗器械通用名称命名规则(国家食品药品监督管理总局令第19号)确定.骨科手术器械的名称可按作用对象和预期用途等方式来命名,如椎管铲刀、颈椎刮匙、椎板咬骨钳、无菌骨牵引针.由网电源、电池或气源提供所需机械动力的骨科手术器械,可命名结构为:电动/电池式+刀具对象+刀具名称,如电动胸骨锯、电动骨钻.骨科手术器械包类产品以体现产品组成、功能用途为基本原则,由多种类别器械组成,一般应以其主要预期用途来命名,如椎间融合手术器械包.(二)产品的结构组成骨科手术器械产品应明确产品结构组成,宜采用图示方式表述产品各组成部分,并明确所用材料.由网电源、电池或气源提供所需机械动力的骨科手术器械一般由单一或多件与配合使用的刀具组成(如:钻类、铣类、磨类、锉类、锯类、刨类).骨科手术器械包类产品由多组件器械产品组成,应明确每个组件的名称.组件可以是外购的具有医疗器械注册证的产品,也可以是尚未注册的医疗器械产品,但其安全有效性要求应与单独注册该组件基本一致.常见产品如图:椎管铰刀椎管刮刀方形铰刀圆形铰刀有齿刮匙弯头刮匙髓核钳颈椎咬骨钳钉孔钻开髓钻平台钻髌骨钻软钻摆锯(三)产品工作原理骨科手术器械产品主要应用机械力学原理.在骨科手术中,与电动/气动/手动工具配合使用,对骨组织实施钻、铣、磨、锯、锉、螺钉打入、螺钉取出、剥离椎骨、修整骨骼或对椎间软组织进行咬切等操作.(四)注册单元划分的原则和实例注册单元划分应根据医疗器械注册管理办法(国家食品药品监督管理总局令第4号)要求,医疗器械注册单元原则上以产品的技术原理、结构组成、性能指标和适用范围为划分依据.骨科手术器械产品不同规格、型号可为同一注册单元.无菌形式和非无菌形式提供的骨科手术器械产品可划为同一注册单元.骨科手术器械包注册单元的划分应首先考虑预期用途,预期用途不同的手术包不作为同一单元.(五)产品适用的相关标准骨科手术器械产品应根据自身特点适用以下标准,但不限于引用以下标准:表1相关产品标准上述标准为本产品所涉及到的相关标准,各企业可以根据实际情况选择引用.如有新版国家标准、行业标准发布实施,应执行最新版本的国家标准、行业标准.(六)产品的适用范围骨科手术器械产品应按照产品使用部位及预期的使用功能来描述产品的适用范围,具体产品举例如下:椎管铲刀:供脊柱手术时铲除骨片及修正骨骼用.椎板咬骨钳:供骨科手术时咬切腐死骨和整修骨骼用.髓核钳:供骨科手术中咬切髓核等软组织用.骨科电动/气动工具配件-刀片、磨头:与气动或电动工具配合使用,供骨科手术中骨的切割、雕塑等用.椎体融合器安装器械包:供骨科手术时安装椎体融合器用.(七)产品的主要风险根据YY/T 0316-2008医疗器械风险管理对医疗器械的应用附录E对骨科手术器械产品已知或可预见的风险进行判定,在进行风险分析时至少应包括以下的主要危害.企业还应根据产品自身特点确定其他危害(见表2),并采取应对措施,确保风险降到可接收程度.表2 产品主要危害(八)产品技术要求的主要性能指标本条给出骨科手术器械需要满足的性能要求,其他性能要求企业可参考相应的国家标准、行业标准,根据产品自身的技术特点制定相应的要求,但不得低于相关强制性国家标准、行业标准的有关要求.企业在制定产品技术要求时,应明确规格型号的划分,且性能指标应至少满足以下要求:1.材料要求骨科手术器械产品应明确产品的材料要求.产品组件的材料牌号及材料所符合的国家标准、行业标准、国际标准,材料牌号的描述应与其符合的标准一致.如不锈钢材料应符合GB/T 1220或YY/T 或GB 4234或ASTM A276-08a等标准的要求;钛合金材料应符合GB/T 10或GB/T 2965或GB/T 3621或GB/T 3623的要求等;铝合金材料应符合GB/T 3190的要求等.产品的材料若无相应的国家标准、行业标准应由供方提供相应的材质证明,应含有基本理化性能等信息.如咬骨钳的头部可选用GB/T 1220中规定的30Cr13,32Cr13Mo,40Cr13材料,也可选用性能优于上述材料的材料.2.硬度骨科手术器械产品硬度如有国家标准、行业标准要求,应执行相应标准(如:YY 1122、YY/T 1127、YY/T 1135、YY 1等);如无相应标准要求,企业可根据产品实际情况明确硬度要求,但应满足临床使用要求.如:咬骨钳(剪)热处理后的头部硬度为:30Cr13材料47 HRC~53 HRC,32Cr13Mo材料48 HRC~56 HRC,40Cr13材料50HRC~58 HRC,左、右两片头部硬度之差应不大于4HRC;电池提供动力的骨科手术器械刀具硬度不低于HRC30;网电源提供动力的骨科手术器械刀具硬度为:不锈钢材硬度不小于650 HV10,其他材料硬度不小于750 HV10,电动片锯齿部表面硬度应不低于HRC 30.3.耐腐蚀性不锈钢材料应具有耐腐蚀性能,不低于YY/T 0149-2006中中b级规定的要求.铝合金制件的阳极氧化膜应符合国家标准GB/T 的规定.4.外观骨科手术器械产品表面应洁净、光滑,无锋棱、毛刺、附着物等缺陷.电池、网电源提供动力的骨科手术器械刀具产品外观应平整,不应有锈迹、锋棱、毛刺和明显麻点; 刃口应无缺口、白口、卷口、裂纹等现象.5.规格尺寸骨科手术器械产品应明确产品规格尺寸,一般采用图表明示.6.使用性能骨科手术器械产品使用性能如有国家标准、行业标准要求,应执行相应标准(如:YY 1122、YY/T 1127、YY/T 1135、YY 1等);如无相应标准要求,企业可根据产品实际情况明确使用性能要求,但应满足临床使用要求.例如:咬骨钳开闭时,腮部与关节应轻松,不得有摆动和卡住现象.咬骨钳闭合后,钳头应互相吻合,无错口现象.咬骨钳鳃轴螺钉应牢固地固定在钳子的一片上,当骨钳开闭时,螺钉不得跟动.7.表面粗糙度产品金属表面粗糙度如有行业标准要求,应按行业标准执行;如无相应行业标准要求,企业可根据产品实际情况明确表面粗糙度要求,但应满足临床使用要求.如:椎板咬骨钳(剪)的表面粗糙度,头部凹槽Ra≤μm,光亮外表面Ra≤μm,无光亮外表面Ra ≤μm;电池提供动力的骨科手术器械刀具表面粗糙度Ra≤μm (刃口除外);网电源提供动力的骨科手术器械刀具表面粗糙度Ra≤μm,刃口部位粗糙度Ra≤μm.8.与有源设备连用的骨科手术器械专用要求(如适用)骨科手术器械产品与电池、网电源、气源动力装置连用时应符合:(1)切削刃磨类刀具网电源提供动力的切削刃磨类刀具,切削刃口应做成右螺旋槽和右切削、刃口沟槽应制成等前角和等螺旋角、基体芯杆圆跳动偏差不大于,切削刃头部直径尺寸公差不大于.电池提供动力的刀具标识应符合YY/T 1052的规定.(2)金钢砂磨类刀具应符合JB/T 中第4章规定的技术要求.(3)锯类刀具应符合YY 1-2005中骨锯通用技术条件的要求.9.无菌提供的产品经确认的方法灭菌后应无菌.若产品经环氧乙烷灭菌,环氧乙烷残留量应不大于10μg/g.10.其他可能涉及的物理要求和化学要求;组件应根据自身特性制定专有技术指标,并能满足使用要求.(九)同一注册单元内注册检验代表产品确定原则和实例同一注册单元中的典型产品是指能够代表本注册单元内其他产品安全性和有效性的产品.骨科手术器械的典型产品应选择能够覆盖注册单元内全部产品性能的产品,若一个型号不能覆盖,除选择典型型号进行全性能检验外,还应选择其他型号进行差异性检验.如同一注册单元中的无菌提供产品与非无菌提供产品,应选无菌提供的产品做为典型型号产品送检;骨科手术器械关键部分,如椎板咬骨钳头部,材料不同应分别送检;电动骨组织手术器械包中,切削刃磨类刀具应与锯类刀具分别送检.(十)产品生产制造相关要求骨科手术器械产品生产企业应进行严格的质量控制.首次注册申报材料应明确产品生产工艺过程,可采用流程图的形式,并提供验证报告说明其过程控制点,如机加工、装配、表面处理、清洗、封口、灭菌(如适用)等工艺过程;应明确表面处理工艺过程中各类加工助剂的添加、去除和残留控制.(十一)产品的临床评价细化要求骨科手术器械产品的临床评价资料应符合医疗器械临床评价技术指导原则的规定.1.对于列入免于进行临床试验的第二类医疗器械目录(国家(以下简称目录)食品药品监督管理总局通告2014第12号附件)及免于进行临床试验的第二类医疗器械目录(第二批)(国家食品药品监督管理总局通告2016年第133号附件)(以下简称第二批目录)中的骨科手术器械产品,临床评价应包括申报产品相关信息与目录、第二批目录所述内容的对比资料,以及申报产品与目录、第二批目录中已获准境内注册医疗器械的对比说明、相应支持性资料.2.对于未列入目录、第二批目录的骨科手术器械产品,可通过同品种医疗器械临床试验或临床使用获得的数据进行分析评价;或按照医疗器械临床评价技术指导原则中的相关要求,开展临床试验进行评价.(十二)产品的不良事件历史记录暂未见相关报道.(十三)产品说明书和标签要求产品说明书和标签的编写要求应符合医疗器械说明书和标签管理规定(国家食品药品监督管理总局令第6号)和医疗器械用于医疗器械标签、标记和提供信息的符号(YY/T )的要求.产品说明书还应包括以下内容:1.应明确非灭菌提供产品使用前的消毒或灭菌方式;2.应明确重复使用产品使用后的清洗方法;3.应注明产品贮存环境要求;4.应明确配套、组合产品使用方法;5.应按照相应行业标准,明确骨科手术器械的标志、标识.(十四)研究资料1.产品性能研究应当提供骨科手术器械性能研究资料以及产品技术要求的研究和编制说明,包括所有指标的确定依据,所采用的标准或方法、采用的原因及理论基础.应明确主要组件的来源及质量要求.2.生物相容性的评价研究骨科手术器械直接与人体组织接触,应按照GB/T16886系列标准对产品进行生物学评价,并提交生物学评价报告证明其安全性.生物相容性评价研究资料应当包括:生物相容性评价的依据和方法;产品所用材料的描述及与人体接触的性质;实施或豁免生物学试验的理由和论证;对于现有数据或试验结果的评价.在进行生物评价过程中,应明确骨科器械产品与人体接触组件或部位的材料性质,如理化性能、生产加工中引入的涂层或阳极氧化等.如采用不锈钢材料生产的咬骨钳产品,根据GB/T 16886系列标准进行生物学评价,应明确其与人体椎体接触部分的材料牌号及要求,阐述与人体椎体接触的部位和时间.生产企业可通过提供咬骨钳产品所用不锈钢材料与上市产品的等同性评价材料,明确选择或放弃生物学试验的理由,汇总已有的数据进行论证,完成生物学评价.3.产品有效期/使用期限和包装研究产品无菌有效期是指灭菌器械能够发挥拟定作用的时间段,无菌有效期验证试验可采用加速稳定性试验和实时稳定性试验,企业需在试验方案中设定检测项目、检测方法及判定标准.产品无菌有效期验证资料可包括以下内容:产品原材料/组件、包装材料、生产工艺、灭菌方法、储存运输条件等基本信息,有效期相关影响因素的说明,稳定性试验的试验方案及试验报告等,包装封口工艺验证方案及报告等.骨科手术器械的有效期可根据产品提供的形式进行考虑,一般无菌方式提供的产品要考虑无菌屏障效期,可与无菌包装验证研究共同进行.产品包装主要对产品起到防护和无菌屏障的作用.以无菌形式提供的产品,其包装验证可依据有关国内、国际标准进行(如GB/T 19633等),提交产品的包装验证报告.包装材料的选择应至少考虑以下因素:包装材料的物理化学性能;包装材料与产品的适应性;包装材料与成型和密封过程的适应性;包装材料与灭菌过程的适应性;包装材料所能提供的物理、化学和微生物屏障保护;包装材料与贮存运输过程的适合性.以非无菌形式提供的产品应考虑包装材料对产品的防护性能及相关要求.三、审查关注点(一)骨科手术器械主要技术性能是否执行了国家和行业的强制性标准;性能指标的确定是否能满足产品的安全有效性;材料要求、硬度要求、外观要求、尺寸要求、使用性能、表面粗糙度要求等是否做出了要求.应明确产品组件原材料牌号要求或供方出厂报告要求;产品组件硬度要求是否符合相关国家标准、行业标准要求或满足临床使用要求.(二)说明书中必须告知用户的信息是否完整,如应明确本产品使用说明、注意事项及警示说明.(三)产品的主要风险是否已经列举,并通过风险控制措施使产品的安全性在合理可接受的程度之内.(四)应关注注册检测产品是否能够代表本注册单元内其他产品安全性和有效性.骨科手术器械(二类)产品技术审评规范编制说明一、指导原则起草目的(一)本指导原则编写的目的是用于指导和规范骨科手术器械产品注册申报过程中审查人员对注册材料的技术审评.(二)本指导原则旨在让初次接触该类产品的注册审查人员对产品原理、结构、主要性能、预期用途等各个方面有个基本了解,同时让技术审查人员在产品注册技术审评时把握基本的尺度,对产品安全性、有效性作出系统评价.二、指导原则编写的依据(一)医疗器械监督管理条例(国务院令第650号)(二)医疗器械注册管理办法(国家食品药品监督管理总局令第4号)(三)医疗器械说明书和标签管理规定(国家食品药品监督管理总局令第6号)(四)医疗器械通用名称命名规则(国家食品药品监督管理总局令第19号)(五)关于发布医疗器械产品技术要求编写指导原则的通告(国家食品药品监督管理总局通告2014年第9号)(六)食品药品监管总局关于实施医疗器械注册管理办法和体外诊断试剂注册管理办法有关事项的通知(食药监械管[2014]144号)(七)关于发布免于进行临床试验的第二类医疗器械目录的通告(国家食品药品监督管理总局通告2014年第12号)(八)关于发布第二批免于进行临床试验医疗器械目录的通告(国家食品药品监督管理总局通告2016年第133号)(九)国家食品药品监督管理部门发布的其他规范性文件三、指导原则中部分具体内容的编写考虑(一)根据医疗器械分类规则确定了骨科手术器械(第二类)产品的分类及适用范围.(二)骨科手术器械命名除参照分类界定通知等相关文件外,主要考虑了临床使用的常见名称和已批准上市产品的名称.(三)本审评规范在编写过程中,正处于医疗器械产品分类调整阶段,新版的医疗器械分类目录还未正式颁布.如新版分类目录颁布后,审评人员可按照最新的要求采纳本规范的相关内容.(四)目前暂未有行业标准或审评规范等文件针对非无菌提供的骨科手术器械使用寿命做出明确规定.依据医疗器械说明书和标签管理规定应明确产品的有效期或使用期限.企业在进行产品效期验证时应考虑产品材料、使用频率、清洗消毒方法对产品的影响等因素,结合实际临床使用的情况,获得有效期或使用期限的验证结果.四、产品技术审评规范编写人员本规范的编写成员由注册行政审批人员、技术审评人员、医疗器械检验人员、临床骨科专家、专业厂家代表等共同组成,以充分利用各方面的信息资源和专业特长,将产品注册各个环节的有关要求尽可能全面地体现在产品技术审评规范的相关章节中,从而保证审评规范的正确、全面、实用.。

广东省卫生厅办公室转发卫生部办公厅关于印发人工关节置换技术管理规范(2012年版)的通知
【法规类别】机关工作综合规定
【发文字号】粤卫办函[2012]404号
【发布部门】广东省卫生厅
【发布日期】2012.09.28
【实施日期】2013.01.01
【时效性】现行有效
【效力级别】XP10
广东省卫生厅办公室转发卫生部办公厅关于印发人工关节置换技术管理规范(2012年
版)的通知
(粤卫办函〔2012〕404号)
各地级以上市卫生局、深圳市卫生和人口计划生育委员会,佛山市顺德区人口和卫生药品监督局,部属、省属驻穗医药院校附属医院及厅直属各医院:
现将《卫生部办公厅关于印发人工髋关节置换技术管理规范(2012版)的通知》(卫办医政发〔2012〕68号)、《卫生部办公厅关于印发人工膝关节置换技术管理规范(2012年版)的通知》(卫办医政发〔2012〕93号)和《卫生部办公厅关于人工关节置换技术管理的补充通知》
1 / 1。



手术动力设备医疗器械产品注册技术审查指导原则一、适用范围本指导原则适用于《医疗器械分类目录》中第二类矫形外科(骨科)手术器械产品中涉及的矫形(骨科)外科用有源器械。
该产品管理类代号为6810-7。
本指导原则适用于由网电源、电池或特定电源为手术刀具(如钻、铣、锯、磨、刨……)提供所需机械动力,在外科或骨科手术中对生物体骨组织的切除处理(如:钻孔、铣削、锯切、磨削……)以及在外科、骨科或耳鼻喉科、整容手术中对生物体骨组织和软组织的刨削处理的非治疗类手术动力设备。
本指导原则不适用于:配备气动装置的骨动力手机设备和牙科的同类设备。
二、技术审查要点(一)产品名称的要求产品命名可采用《医疗器械分类目录》中的名称,命名结构为:电动/电池式+刀具对象+刀具名称。
例如:电动胸骨锯、电动骨钻、电动石膏剪、电动石膏锯、电池式自停颅骨钻。
带有多种刀具的设备建议直接采用行业标准YY/T 0752-2009中的通用名称:电动骨组织手术设备。
在实际应用中常采用的产品名称有:手术动力装置、手术动力设备、手术动力系统、电动动力系统、充电式电动骨科手术设备、电池动力系统、骨动力系统等。
(二)产品的结构和组成1.产品的结构和组成(1)电网供电的手术动力设备结构A:由控制单元、软轴动力传输单元、输出机械力驱动的手机、刀具、附属附件(部件)等组成。
结构B:由控制单元、电缆、输出机械力驱动的手机、刀具、附属附件(部件)等组成。
由于以上两种结构的差异性,其性能和适用性各有不同,根据临床的不同功能要求,设备可以单独采用结构A或结构B,也可以采用结构A和结构B的组合。
(2)电池供电手术动力设备由电池供电,由手机、刀具、电池和(或)电池充电器等组成,提供锯类、钻类、磨(锉)类、刀类等骨组织手术刀具所需机械动力实施骨组织手术的医疗器械。
2.组成单元结构/功能描述(1)控制单元为手机提供机械动力能和/或电能,并对其输出实施实时监控的单元,由控制面板和/或脚踏开关对控制单元进行功能选择和切换控制。

骨科手术器械类产品技术审评规范(2012版)根据《医疗器械注册管理办法》(国家食品药品监督管理局令第16号)的要求并结合骨科手术器械类产品的特点,为规范骨科手术器械类产品的技术审评工作,特制定本规范。
一、适用范围本规范适用于无源类常规手术器械,带电手术器械(如电动骨锯)不在此规范涉及范围内。
依据《医疗器械分类目录》,骨科手术器械(以下简称骨科器械)管理类别分为I类和II类,产品类代号为6810。
二、技术审查要点(一)产品名称的要求骨科器械的名称可分为两大类,一类可按作用部位、适用范围、手术方式来命名,一般由多种类别器械组成,如:髋关节手术器械、膝关节手术器械、脊柱内固定手术器械等;另一类可按器械的结构、性能来命名,一般为单一类别器械,如:刀锥类骨科手术器械、剪锯类骨科手术器械等。
如按前种命名,应注重明确产品的各种组件和其具体性能要求;如按后种命名,应注重明确产品的适用范围和产品之间配合使用要求。
(二)产品的结构组成应明确产品的结构和组成。
(三)产品工作原理主要应用机械力学原理。
(四)产品适用的相关标准常用的国家标准、行业标准包括:GB/T 191-2008 包装储运图示标志GB/T 230.1-2009金属材料洛氏硬度试验第1部分:试验方法(A、B、C、D、E、F、G、H、K、N、T标尺) GB/T 1220-2007 不锈钢棒GB/T 4340.1-2009 金属材料维氏硬度试验第1部分:试验方法GB/T 5132.5-2009 电气用热固性树脂工业硬质圆形层压管和棒第5部分:圆形层压模制棒GB 4234-2003 外科植入物用不锈钢GB/T 13810-2007 外科植入物用钛及钛合金加工材GB/T 2965-2007 钛及钛合金棒材GB/T3621-2007 钛及钛合金板材GB/T 3623-2007 钛及钛合金丝YY/T 0149-2006 不锈钢医用器械耐腐蚀性能试验方法YY/T 0294.1-2005 外科器械金属材料第1部分:不锈钢YY/T 1052-2004 手术器械标志YY 1122-2005 咬骨钳(剪)通用技术条件YY/T 1127-2006 咬骨钳YY/T 1135-2008 骨剪YY 1137-2005 骨锯通用技术条件YY 91071~91073-1999 平骨凿圆骨凿医用凿凿切性能测试方法平凿、圆凿YY/T 91141-1999 骨科凿类通用技术条件注:应执行产品适应的标准,并不局限于以上标准。

矫形器产品技术审评规范(2012版)根据《医疗器械注册管理办法》(国家食品药品监督管理局令第16号)的要求并结合矫形器产品的特点,为规范该类产品的技术审评工作和指导该类产品的注册申报工作,特制定本规范。
一、适用范围本规范适用于《医疗器械分类目录》中6810的第I类矫形器产品,即上肢类矫形器、下肢类矫形器、脊柱类矫形器。
矫形器是指装配于人体四肢、躯干等部位的体外器具的总称,通过对肢体/躯干某个部位施加影响,达到保护、固定、代偿、矫正的目的。
产品分类见下表:二、技术审查要点(一)产品名称的要求矫形器的产品名称命名要求原则为:□□矫形器产品名称体现功能性(必要时)具体肢体部位举例:膝关节矫形器;脊柱侧弯矫形器。
(二)产品的结构和组成1.典型产品外形结构示意图(1)手指矫形器(2)胸腰椎矫形器(3)下肢矫形器2.产品的结构和组成企业应明确描述产品的具体结构和组成部分。
3.产品的分类(见上表)(三)产品的工作原理矫形器通过对肢体/躯干某个部位施加影响,达到保护、固定、矫正的目的。
(四)产品适用的相关标准GB/T191-2008 包装储运图示标志GB 9174-2008 一般货物包装通用技术条件GB/T 10000-1998 中国成年人人体尺寸GB/T 14191-1993 假肢和矫形器术语GB/T16886.1-2001 医疗器械生物学评价第1部分:评价与试验GB/T16886.5-2003 医疗器械生物学评价第5部分:体外细胞毒性试验GB/T16886.10-2005 医疗器械生物学评价第10部分:刺激与迟发型超敏反应试验MZ 003-1993 支条式下肢矫形器MZ 004-1993 支条式脊柱矫形器YY 0076-1992 金属制件的镀层分类技术条件YY0316-2008 医疗器械-风险管理对医疗器械的应用YY0466-2003 医疗器械用于医疗器械标签、标记和提供信息的符号应执行产品适应的标准,并不局限于以上标准。
医疗器械产品注册技术审评报告引言本报告是针对医疗器械产品进行的技术审评报告,旨在对该产品的技术性能进行评估和审查,以确定其是否符合相关法规和标准的要求。
本报告将对该医疗器械产品的性能特点、临床需求以及技术参数进行详细分析,并提出审评结论和建议。
产品概述该医疗器械产品为一种新型手术器械,主要用于腹腔镜手术中器械的取放。
该产品由不锈钢制成,具有良好的耐腐蚀性和刚性。
产品采用了人体工程学设计,使得手术医生能够更加方便地进行腹腔镜手术操作,提高手术效率并减少手术风险。
技术参数分析1. 外观参数•尺寸:长40cm、宽15cm、高5cm•材质:不锈钢•颜色:银色•外观处理:光滑处理,无明显划痕或变形2. 技术性能参数•手柄设计:符合人体工程学原理,操作舒适、灵活•运动灵活度:产品可在各个方向上灵活转动,轻松适应手术需求•耐腐蚀性:不锈钢材质具有良好的耐腐蚀性,产品可在不同环境下使用•重量:适中的重量,方便手术医生操作并减轻手臂疲劳感性能测试为了评估该医疗器械产品的性能,我们进行了以下测试:1. 手柄设计测试在这个测试中,我们邀请了十位腹腔镜手术专家使用该产品进行操作,通过观察和调查他们的反馈,评估手柄设计的舒适性和灵活性。
结果表明,产品的手柄设计符合人体工程学原理,手术医生普遍认为手柄舒适度高,容易掌握并操作灵活。
2. 运动灵活度测试为了评估产品在不同方向上的运动灵活度,我们进行了一系列动作测试。
结果显示,产品具有良好的运动灵活度,可以在多个角度上灵活转动,方便手术医生进行操作。
3. 耐腐蚀性测试我们将产品暴露在盐水和酸性溶液中,以模拟不同环境条件下的使用情况。
经过一段时间的浸泡后,我们检查产品表面是否发生腐蚀或变色。
测试结果表明,产品具有良好的耐腐蚀性,外观无明显变化。
4. 重量测试我们通过称重量器测量产品的重量,结果显示产品重量在合理范围内。
手术医生们表示产品的重量适中,不会给手臂带来过多负担,能够减轻手术中的疲劳感。
骨科外固定架技术审评规范根据《医疗器械注册管理办法》(国家食品药品监督管理局令第16号)的要求并结合骨科外固定架的特点,为规范该类产品的技术审查工作和指导该类产品的注册申报工作,特制定本规范。
一、适用范围本规范适用于外固定架类医疗器械,该类医疗器械管理类别分类见下表二、技术审查要点(一)产品名称的要求外固定架类医疗器械产品名称应以“作用部位+预期用途”的形式给出。
(通用)(二)产品的结构组成及作用原理外固定架的生产厂家不同,结构也不相同,有简单的,也有复杂的。
但基本结构是可调支架,在临床上需配合接骨螺钉(或骨针)使用。
但本产品不包括接骨螺钉(或骨针)可调支架的结构多种多样,根据使用部位的不同,采取不同的组合方式,将植入人体的接骨螺钉(或骨针)连接起来,使骨折端稳定固定,并逐渐愈合。
(三)产品适用的相关标准YY/T0149-2006 不锈钢医用器械耐腐蚀性能试验方法GB1220-1992 不锈钢棒GB191-2000 包装储运图示标志()产品的预期用途外固定架主要通过骨内穿针来实现畸形矫正,固定、加压或牵拉骨端。
(五)产品的主要风险使用中连接件松动,不稳定。
材料强度不够,发生断裂、变形等。
(六)外固定架的主要技术指标1、主要材料材料应耐消毒、并符合材料的相关标准要求。
(如GB1220、);另外应考虑材料便于清理、清洁。
2、产品外观要求产品表面应光滑、无锋棱、毛刺、附着物等缺陷。
3、金属材料耐腐蚀性能(YY/T0149)通常情况下,在室温下工作,环境比较好(不在潮湿环境中工作)。
只要在清水中浸泡4小时不生锈,正常情况下也不会生锈,满足使用要求。
4、应有明确的规格尺寸要求5、产品各部件应符合组装要求。
(七)产品的检测要求(规定出厂检验)该产品的检测除了主要技术指标外,还应检测产品外观和使用性能。
1、产品外观:表面应光滑,无锋棱、毛刺、附着物等缺陷。
2、使用性能:各活动部件应活动自如、锁紧牢靠,符合使用要求。
骨组织手术设备产品注册技术审查指导原则本指导原则旨在指导和规范第二类骨组织手术设备产品的技术审评工作,帮助审评人员理解和掌握该类产品的原理、结构、性能、预期用途等内容,掌握技术审评工作的基本要求和尺度,对产品安全性、有效性作出系统评价。
本指导原则系对骨组织手术设备注册技术审查的通用要求,申请人/生产企业应依据具体产品的特性对注册申报材料的内容进行充实细化。
申请人/生产企业还应依据具体产品的特性确定其中的具体内容是否适用,若不适用,需详细阐述其理由及相应的科学依据。
本指导原则是对产品的技术审查人员和申请人/生产企业的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在当前认知水平下制订的,随着科学技术的不断发展,随着相关法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整和更新。
审查人员仍需密切关注相关适用标准与注册法规的变化,以确认申报产品是否符合法规要求。
一、适用范围本指导原则适用于《医疗器械分类目录》中第二类矫形外科(骨科)手术器械产品中涉及的矫形(骨科)外科用有源器械。
该产品管理类代号为6810-7。
本指导原则适用于由网电源、电池或特定电源为手术刀具(如钻、铣、锯、磨、刨等)提供所需机械动力,在外科或骨科手术中对生物体组织进行手术处理(如:钻孔、铣削、锯切、磨削等)以及在外科、骨科或耳鼻喉科、整容手术中对生物体硬组织和软组织进行刨削处理的非治疗类骨组织手术设备。
本指导原则不适用于:配备气动装臵的骨动力手机设备和牙科的同类设备。
二、技术审查要点(一)产品名称的要求产品命名可采用《医疗器械分类目录》中的名称,命名结构为:电动/电池式+手术对象+刀具名称。
例如:电动胸骨锯、电动骨钻、电动石膏剪、电动石膏锯、电池式自停颅骨钻。
三级医院骨科复审手术材料清单【手术材料清单】- 三级医院骨科复审尊敬的医生或相关工作人员:为了确保手术的顺利进行以及患者的安全,我们提前准备了以下骨科复审手术材料清单。
请您在手术前仔细核对,确保所有材料的准备完整与规范,以确保手术的成功进行。
一、器械类1. 手术台:确保手术台稳固可靠,具备调节高度和角度等功能,以满足不同手术需求。
2. 手术椅和踏板:用于医生和护士的操作,确保操作人员的舒适度和操作的便利性。
3. 手术灯:提供充足明亮的光源,确保手术区域的清晰可见。
4. 手术器械:包括但不限于骨锁板、骨钳、骨钻、骨锤、骨锯等骨科手术所需的常用器械。
5. 手术钢板和螺钉:根据手术需求选择不同材质和规格的手术钢板及螺钉,以保证手术的稳定性和成功性。
6. 一次性物品:如一次性手术刀片、手术纱布、手术膜等,确保手术过程的无菌性和安全性。
二、消毒清洁类1. 消毒液:如乙醇、碘伏、氯己定等,用于器械和手术区域的消毒清洁,确保术中环境的无菌。
2. 消毒器械及容器:如消毒腔、器械槽等,用于容纳和消毒手术器械,确保器械的无菌和使用安全。
3. 无菌巾:用于擦拭手术台、手术器械等,保持手术区域的清洁和无菌。
4. 手术器械清洗剂:用于清洗手术器械,保证手术器械的清洁度和无菌。
5. 手套、口罩和帽子:用于手术人员的个人防护,防止交叉感染和手术区域的二次污染。
三、麻醉药物类1. 麻醉剂:如丙泊酚、吗啡、芬太尼等,根据患者情况和手术的需要,选择合适的麻醉药物。
2. 麻醉辅助药物:如异丙酚、氧化亚氮等,用于辅助麻醉的药物。
3. 镇痛药物:如吗啡、可待因等,为手术后患者提供镇痛效果。
四、医用耗材类1. 手术服和手术巾:包括无菌手术服、手术帽、手术袜等,用于手术人员和患者的穿戴,保持手术环境的无菌。
2. 纱布和绷带:用于手术伤口的包扎与敷料,防止感染和促进伤口的愈合。
3. 注射器和针头:用于药物的注射和静脉通道的开通。
4. 引流管:用于排除手术中产生的血液和组织液,保持手术区域的干燥与清洁。
附件1骨科外固定支架注册技术审查指导原则(2018年修订)本指导原则旨在为申请人进行骨科外固定支架注册申报提供技术指导,同时也为药品监督管理部门对注册申报资料的审评提供技术参考。
本指导原则是对骨科外固定支架注册申报资料的一般要求,申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
一、适用范围本指导原则适用于《医疗器械分类目录》中按照第二类医疗器械管理的骨科外固定支架。
—1 —本指导原则不包含与其配套使用的金属骨针产品。
本指导原则不包含矫形用外固定支架。
矫形用外固定支架及骨融合术后辅助固定用外固定支架产品可参考本指导原则进行注册申报。
二、技术审查要点(一)产品名称要求骨科外固定支架应符合《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)的要求,可采用相关国家标准、行业标准上的通用名称,或以产品结构和适用范围为依据命名。
例如:一体式骨科外固定支架、组合式骨科外固定支架等。
(二)产品的结构和组成1.产品的结构和组成外固定支架按结构组成可分为一体式和组合式两种类型。
按照交付状态可分为非无菌形式提供和无菌形式提供两种。
一体式外固定支架主要由横(竖)针夹、延长接头、撑开架、偏心轴、万向球等部件组成。
按照结构组成可分为多种型式,每种型式按照使用部位又可分为多种型号。
组合式外固定支架可根据使用部位由闭合环、开放环、环针夹、环杆(管)夹、直形杆(管)等组成。
受理号:CQZ1600326医疗器械产品注册技术审评报告(境内)产品中文名称:可吸收硬脑膜封合医用胶产品管理类别:第三类申请人名称:山东赛克赛斯药业科技有限公司国家食品药品监督管理总局医疗器械技术审评中心目录基本信息 (3)产品审评摘要 (4)一、产品概述 (4)二、临床前研究摘要 (6)三、临床评价摘要 (10)四、风险分析及说明书提示 (12)综合评价意见 (15)基本信息一、申请人名称:山东赛克赛斯药业科技有限公司二、申请人住所:济南市高新区开拓路2222号三、生产地址:济南市高新区开拓路2222号产品审评摘要一、产品概述(一)产品结构及组成可吸收硬脑膜封合医用胶(以下可简称“医用胶”)由粉剂、溶液(溶液A、溶液B)和配套工具组成,其中粉剂的成分为分子量20000左右的四臂聚乙二醇-N-羟基丁二酰亚胺-戊二酸酯(4-arm-PEG-SG)、染色剂亮蓝85和抗氧化剂二丁基羟基甲苯(BHT);溶液A为酸性磷酸盐缓冲溶液;溶液B为含三赖氨酸(Tri-lys)和聚乙烯亚胺(PEI)的碱性四硼酸钠缓冲溶液;配套工具包括双联注射器架、混液连接配件、混液喷头。
产品组成的示意图如图1所示。
图1 产品组成示意图该产品中4-arm-PEG-SG装于粉剂瓶中,溶液A和溶液B装于注射器中。
配套工具经环氧乙烷灭菌后与产品一起包装,整体经辐射灭菌,一次性使用。
(二)产品适用范围该产品适用于开颅手术中,硬脑膜缝合部位的辅助封合,防止脑脊液渗漏。
(三)型号/规格该产品包装规格为2mL、3mL、4mL、5mL、6mL。
各包装规格的组成如表1所示。
表1 产品规格(四)作用机理该产品主要组分4-arm-PEG-SG、Tri-lys和PEI在混合时发生亲核取代反应,形成以酰胺键结合的交联大分子,即水凝胶。
水凝胶的粘合作用一方面来自水凝胶内部分子以酰胺键交联键合的作用力,另一方面是改性聚乙二醇与施用部位组织蛋白质的氨基发生同样亲核取代反应的键合力。
骨科手术器械类产品技术审评规范(2012版)
根据《医疗器械注册管理办法》(国家食品药品监督管理局令第16号)的要求并结合骨科手术器械类产品的特点,为规范骨科手术器械类产品的技术审评工作,特制定本规范。
一、适用范围
本规范适用于无源类常规手术器械,带电手术器械(如电动骨锯)不在此规范涉及范围内。
依据《医疗器械分类目录》,骨科手术器械(以下简称骨科器械)管理类别分为I类和II类,产品类代号为6810。
二、技术审查要点
(一)产品名称的要求
骨科器械的名称可分为两大类,一类可按作用部位、适用范围、手术方式来命名,一般由多种类别器械组成,如:髋关节手术器械、膝关节手术器械、脊柱内固定手术器械等;另一类可按器械的结构、性能来命名,一般为单一类别器械,如:刀锥类骨科手术器械、剪锯类骨科手术器械等。
如按前种命名,应注重明确产品的各种组件和其具体性能要求;如按后种命名,应注重明确产品的适用范围和产品之间配合使用要求。
(二)产品的结构组成
应明确产品的结构和组成。
(三)产品工作原理
主要应用机械力学原理。
(四)产品适用的相关标准
常用的国家标准、行业标准包括:
GB/T 191-2008 包装储运图示标志
GB/T 230.1-2009金属材料洛氏硬度试验第1部分:试验方法(A、B、C、D、E、F、G、H、K、N、T标尺)
GB/T 1220-2007 不锈钢棒
GB/T 4340.1-2009 金属材料维氏硬度试验第1部分:试验方法
GB/T 5132.5-2009 电气用热固性树脂工业硬质圆形层压管和棒第5部分:圆形层压模制棒
GB 4234-2003 外科植入物用不锈钢
GB/T 13810-2007 外科植入物用钛及钛合金加工材
GB/T 2965-2007 钛及钛合金棒材
GB/T3621-2007 钛及钛合金板材
GB/T 3623-2007 钛及钛合金丝
YY/T 0149-2006 不锈钢医用器械耐腐蚀性能试验方法
YY/T 0294.1-2005 外科器械金属材料第1部分:不锈钢YY/T 1052-2004 手术器械标志
YY 1122-2005 咬骨钳(剪)通用技术条件
YY/T 1127-2006 咬骨钳
YY/T 1135-2008 骨剪
YY 1137-2005 骨锯通用技术条件
YY 91071~91073-1999 平骨凿圆骨凿医用凿凿切性能测试方法平凿、圆凿
YY/T 91141-1999 骨科凿类通用技术条件
注:应执行产品适应的标准,并不局限于以上标准。
(五)产品的预期用途
产品预期用途范例如下:
剪类:用于剪断骨、韧带、软组织或器械
钛网剪 钢丝剪
锯类:用于锯断骨。
骨锯
钩类:用于显露手术视野的各种拉钩。
骨钩
刮类:刮勺:刮除病灶用;骨膜剥离子:用于剥离骨膜;扩孔。
刮匙
骨膜剥离子
钳类:对骨的去除、夹持、撑开或压缩、或对植入物的塑形等。
持板钳 弯棒钳
钻、锥类:用于在骨上打孔、攻丝。
丝锥 丝锥
刀类:用于切除、截断骨。
骨刀
辅助工具类:导向器:作为骨科器械配套工具,起导向和组织保护作用;测深尺:主要用于测量孔的深度;持钉镊:用来夹取骨螺钉;打入器:用于打入骨科植入物;试模:用于临时复制骨形状;钢板弯曲扳手:用于接骨板折弯;紧固工具:用于紧固或卸下螺钉、螺母。
导向器测深尺
持钉镊打入器
试模钢板弯曲扳手
改锥
组合工具类:一般用于某项特定的临床用途,如髋关节置换手术工具等。
(六)产品的主要风险
骨科器械一般应考虑产品的材质、消毒要求和产品结构造成的不易清洗,电动骨钻、骨锯等有源器械还应考虑电气安全风险。
(七)产品标准的主要技术要求
骨科器械产品的主要技术要求一般包括以下几项:
1.材料
应选择耐高温、无毒、不易生锈的材料。
如:咬骨钳(剪)的头部可选符合GB/T 1220规定的32Cr13Mo、30Cr13、40Cr13等材料,也可选用性能优于上述材料的材料;骨锯的锯片可采用GB/T 1220中规定材料及65Mn、60SiMn等材料制造;骨凿可采用GB/T 1220中规定的40Cr13和90Cr18MoV 等材料制造。
其它不锈钢材料应符合GB/T 1220或YY/T 0294.1或GB 4234的要求。
非金属材料:聚乙烯、硅胶、聚甲醛、聚四氟乙烯、夹布胶木等。
钛合金材料应符合GB/T 13810或GB/T 2965或GB/T 3621或GB/T 3623的要求。
材质证明(如有):如适用可包含化学成分、力学性能(可包含抗拉、屈服、延伸、抗弯等性能)等。
其它材料适用于手术器械的非主体部分时,应能满足高温高压灭菌要求。
以上为目前常用于骨科手术器械的材料及标准,本规范并不局限于以上要求,只要能满足临床使用需求及国家的相关法规文件,也可用于骨科手术器械的生产。
2.硬度
产品硬度如有行业标准要求,应执行行标(如:YY 1122、YY/T 1127、YY/T 1135、YY 1137等);如无相应行标要求,企业可根据产品实际情况明确硬度要求,但应满足临床使用要求。
如:咬骨钳(剪)热处理后的头部硬度为:30Cr13材料47 HRC~53 HRC,32Cr13Mo材料48 HRC~56 HRC,40Cr13材料50HRC~58 HRC,左、右两片头部硬度之差应不大于4HRC;片锯齿部表面硬度应不低于30HRC;骨凿热处理后的硬度为:头部20
毫米内:40Cr13材料50HRC~55 HRC,90Cr18MoV材料53HRC~58 HRC,其余部位不低于35HRC。
3.耐腐蚀性(如产品采用不锈钢为主体材料)
应耐腐蚀,根据制造材料的不同、器械的用途不同,选择YY/T 0149中规定的试验方法的相应评价方法。
4.外观
表面应洁净、光滑,无锋棱、毛刺、附着物等缺陷。
5.规格尺寸
应明确产品规格尺寸和公差。
(可采用图表明示)
6.使用性能
各活动部件应活动自如,并应能满足使用要求。
7.表面粗糙度
产品表面粗糙度如有行业标准要求,应按行标执行;如无相应行标要求,企业可根据产品实际情况明确表面粗糙度要求,但应满足临床使用要求。
如:咬骨钳(剪)的表面粗糙度:头部凹槽Ra≤0.8μm,光亮外表面Ra≤0.4μm,无光亮外表面Ra≤0.8μm;骨锯的锯片Ra≤0.8μm,齿部Ra≤6.3μm,手柄Ra≤1.6μm。
8.组件要求(如有)
如产品由多个部件组成,还应对各组件有相应具体要求。
(八)产品的临床试验要求
需要进行临床试验时,应符合《医疗器械临床试验管理规定》(国家局5号令)的要求。
(九)产品说明书、标签、包装标识
产品说明书、标签、包装标识应按照国家局10号令的相关要求。
产品上的永久性标识按YY/T1052制作。
产品说明书应包括如下内容:
1.应明确产品使用前的消毒或灭菌方式;
2.应明确产品使用后的清洗方法;
3.应注明产品贮存环境要求;
4.应明确配套、组合产品使用方法。
(十)注册单元的划分
产品注册单元的划分按以下原则:
1.按同一手术类型划分(临床专家)
例:髋关节手术器械、膝关节手术器械等;
2.按性能、用途划分
例:刀类、锥类、剪类等。
如按照第二种划分原则,应符合《医疗器械分类目录》要求。
(十一)同一注册单元中典型产品的确定原则
典型产品的确定主要依据器械的材料、结构、形状复杂程度、尺寸大小、长径比、加工难度、使用量等因素。
(十二)常用的生产和检测设备
常用生产设备一般包括车床、铣床、磨床、钻床等;
常用检测设备一般包括硬度计、千分尺、百分表、卡尺、粗糙度样块、耐腐蚀试验设备等。
骨科手术器械类产品技术审评规范修订说明
本规范于2008年3月首次发布。
三年来,通过日常审批、生产企业的反馈,我们获得了一些有价值的意见和建议。
为了更加有效的实施此规范,我们对规范进行第二次修订,现将修订内容说明如下:
1.更新最新版本的相关引用标准。
2.调整第五部分产品适用的相关标准,增加部分可能引用
到的标准。
3.调整钳类、剪类和刮类手术器械的适用范围。
4.增加非金属材料,钛合金材料的材质要求,提出了对其
他材料应用于手术器械非主体部分要求。
5.调整产品表面粗糙度要求。
6.调整了产品的硬度要求,明确了产品硬度可参考的标
准。
7.由于GB1220目前已有最新版本,不锈钢牌号有所变化,
本规范对此部分内容进行了调整。