红外分光光度法
- 格式:doc
- 大小:49.00 KB
- 文档页数:5



红外分光光度法红外分光光度法 §1 概述UV 光谱又称:电子光谱、振一转光谱一、定义:由分子的振动—转动能级跃迁产生的光谱为红外光谱。
是以速光率T —ζ或T —λ图来进行定性、定量分析方法。
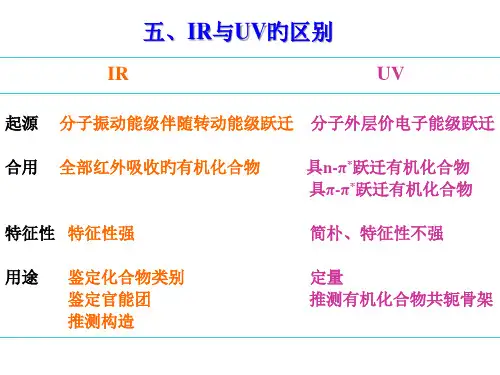
T-λ曲线,由于波长等距,曲线“前密后疏” 使用较多的是T-ζT-ζ曲线,由于波数等距,曲线“前疏后密” 使用较多的是T-ζ 二、IR 与UV 的区别:1、起源不同:UV 是分子的价电子跃迁产生(电子光谱)IR 是分子振-转能级跃迁产生(振-转光谱)2、适用范围:UV 主要讨论芳香化合物、共轭、长共轭化合物,且限于溶液。
IR :几乎所有的有机化合物,且用于固、液、气。
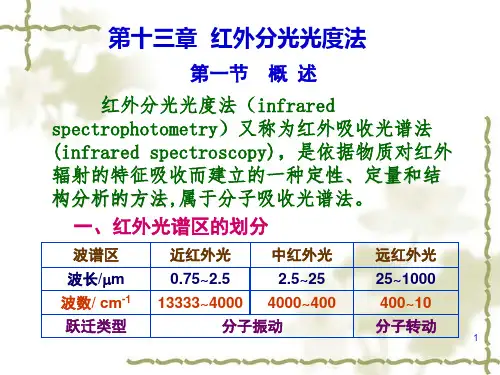
3、特征性强:ζ波数cm -1=)(104m μλ0.76~2.5μm 近红外 2.5~25μm 中红外(中红外研究最为)4000~400 25~500μm 定红外红外主要用于定性、紫外主要用于定量。
三、用途:定性、定量、定结构构型、取代基位置定结构:①官能量;②化学类别;③精细结构 直链、支链 §2 基本原理IR :由峰位、峰形、峰强描述主要讨论:起源、峰位、峰形、峰强及其影响因素。
一、振动能级和振动光谱讨论双原子分子。
re-平衡位置时原子间距。
将A 、B 两原子看作两个小球,化学键质量可忽略,则两个原子沿键轴方向的伸缩振动可近似为简谐振动,双原子分子视为谐振子。
1、位能:U=2)(21re r K -r=re, U=0r>re, U>0 r<re, U>0由量子力学可推,分子振动的总能量:Ev=(V+Ev=(V+21)h υ V=0,1,2……(振动量子数)由Hu 克定律:F=-Kr ,条件:弹簧伸长量不能太大。
而由图15-4,V=0时,E V =21h υ 振幅小V=1时,E V =23h υ ……振幅增大当大到一定程度时,化学键断裂了,即分子离解了(达到了离解能) ∴真实分子并不是谐振 2、基频峰产生的重要条件 V=0 V=1由图15-4,振动能级是量子化的,分子只能吸收相当于两个能级差的光量子,才能发生跃迁。




第4章 红外分光光度法一、内容提要红外线(infrared ray )是波长为0.76~1000μm 的电磁波。
红外分光光度法(infrared spectrophotometry )是依据物质对红外辐射的特征吸收而建立的一种分析方法,即红外光谱法。
红外分为近、中、远三个区域,通常红外光谱指中红外吸收光谱,由分子中原子的振动能级跃迁和分子的转动能级跃迁所产生的光谱,故为振动-转动光谱,简称振-转光谱。
红外吸收光谱又称红外吸收曲线,多用透光率-波数(T -σ)曲线描述,所谓吸收峰实际上是曲线的“谷”。
一条红外吸收曲线的特征主要由吸收峰的位置(λmax 、σmax )、吸收峰的个数及吸收峰的强度来描述。
分子吸收适当频率的红外辐射(L νh )后,可以由基态跃迁至激发态,其所吸收的光子能量必须等于分子振动能量之差,即ννh E h V V L ∆=∆=,即ννV L ∆= 或σσV L ∆= 是产生红外吸收峰的必要条件之一。
双原子分子只有一类振动形式(mode of vibration )为伸缩振动。
多原子分子有两类振动形式为伸缩振动和弯曲振动。
振动自由度(f )是分子基本振动的数目,非线性分子, 63-=N f ;线性分子,53-=N f 。
在红外光谱中,某一基团或分子的基本振动能吸收红外线而发生能级跃迁,必须满足两个基本条件:(1)振动过程中,△μ≠0;(2)必须服从 νL =ΔV ν,两者缺一不可。
泛频峰使吸收峰数多于基本振动数,简并和红外非活性振动使基频峰数少于基本振动数。
吸收峰的位置或称峰位通常用σmax (或νmax 、λmax )表示,对基频峰而言,σmax =σ,基频峰的峰位即是基团或分子的基本振动频率。
μk σ'=1307(cm -1) 折合相对质量相同时,化学键力常数越大,则基本振动频率越大。
化学键相同时,随着折合相对质量μ'的增大,其吸收频率变低。
吸收峰峰位由化学键两端的原子质量和化学键的键力常数预测,在比较复杂的分子中,由于有诱导效应(induction effection ,I 效应)、共轭效应(conjugation effect ,M 效应)、空间位阻、氢键等因素影响,峰位产生10~100cm -1的位移。

分析化学基础知识——第七课红外分光光度法第七课红外分光光度法⼀、概述1.红外区波长范围及分区波长范围:0.76µm-1000µm分区:2.红外吸收光谱的表⽰⽅法3.IR的特点适⽤于⽓、液、固态样品、且样品⽤量少。
⼤多数化合物均有红外吸收,除了单原⼦分⼦和同核分⼦。
红外光谱中的吸收峰较多,特征性强,适合⽤于定性和结构解析。
红外光谱仪的价格相对低廉。
定量分析灵敏度差,准确度低,主要⽤于定性分析。
不适合作含⽔样品的分析。
⼆、基本原理分⼦振动和红外吸收吸收峰的位置吸收峰的强度1.分⼦振动和红外吸收双原⼦分⼦的振动与红外吸收分⼦振动简单的双原⼦A-B间的振动可近似地⽤谐振⼦模型来描述振动频率可由虎克定律和⽜顿定律推导出来A、B视为两个刚性⼩球化学键视为质量忽略不计的弹簧A、B间的振动视为简谐振动红外吸收⼊射光频率与分⼦振动频率相等时,分⼦将吸收⼊射光,振动振幅加⼤,产⽣吸收光谱,因此,所吸收光的频率为:多原⼦分⼦振动形式伸缩振动γ弯曲振动δ(1)伸缩振动键长变化但键⾓不变的振动它包括两种类型对称伸缩振动γs反称伸缩振动γas亚甲基的伸缩振动(2)弯曲振动键⾓发⽣周期性变化,但键长不变的振动。
它包括以下⼏种类型⾯内弯曲振动 AX2⾯外弯曲振动变形振动AX3⾯内弯曲振动(β)剪式振动(δ)⾯内摇摆振动(ρ)⾯外弯曲振动(γ)⾯外摇摆振动(ω)扭曲振动(τ)变形振动对称变形振动(δs)不对称变形振动(δas)(3)振动⾃由度双原⼦分⼦:⼀种振动形式多原⼦分⼦:振动形式复杂,可以分解为许多简单的基本振动。
基本振动的数⽬称为振动⾃由度,可以⽤作估计基频峰的可能数⽬。
振动⾃由度的计算分⼦的运动形式分为:平动、振动和转动,则:振动⾃由度=总⾃由度-平动⾃由度-转动⾃由度设:分⼦含有N个原⼦则:总⾃由度为3N,平动⾃由度为3转动⾃由度为3(对于⾮线形分⼦)或2(对于线形分⼦)振动⾃由度⾮线形分⼦线形分⼦3N-6 3N-5H2O分⼦的振动⾃由度3×3-6=3CO2的振动⾃由度3×3-5=4基频峰数⽬与振动⾃由度通常,基频峰数⽬<振动⾃由度原因:简并红外⾮活性振动仪器的分辨率或灵敏度不够⾼产⽣红外吸收峰的条件1.辐射恰好提供物质产⽣振动跃迁所需的能量。

红外分光光度法1 简述红外分光光度法是在4000~400cm -1波数范围内测定物质的吸收光谱,用于化合物的鉴别、检查或含量测定的方法,化合物受红外辐射照射后,使分子的振动和转动运动由较低能级向较高能级跃迁,从而导致对特定频率红外辐射的选择性吸收,形成特征性很强的红外吸收光谱,红外光谱又称振—转光谱。
红外光谱是鉴别物质和分析物质化学结构的有效手段,已被广泛应用于物质的定性鉴别、物相分析和定量测定,并用于研究分子间和分子内部的相互作用。
习惯上,往往把红外区分为3个区域,即近红外区(12800~4000cm -1,0.78~2.5µm),中红外区(4000~400cm -1,2.5~25µm)和远红外区(400~10cm -1,25~1000µm)。
其中中红外区是药物分析中最常用的区域。
红外吸收与物质浓度的关系在一定范围内服从于朗伯—比尔定律,因而它也是红外分光光度法定量的基础。
红外分光光度计分为色散型和傅里叶变换型两种。
前者主要由光源、单色器(通常为光栅)、样品室、检测器、记录仪、控制和数据处理系统组成。
以光栅为色散元件的红外分光光度计,波数为线性刻度,以棱镜为色散元件的仪器以波长为线性刻度。
波数与波长的换算关系如下:)(10)41m cm μ波长波数(=-傅里叶变换型红外光谱仪(简称FT-IR )则由光学台(包括光源、干涉仪、样品室和检测器)、记录装置和数据处理系统组成,由干涉图变为红外光谱需经快速傅里叶变换。
该型仪器现已成为最常用的仪器。
2 红外分光光度计的检定所用仪器应按现行国家质量与核查技术监督局“色散型红外分光光度计检定规程”、“傅里叶变换红外光谱仪检定规程”和《中国药典》2015年版四部通则0401规定,并参考仪器说明书,对仪器定期进行校正规定。
2.1 波数准确度2.1.1 波数准确度的允差范围 傅里叶变换红外光谱仪在3000cm -1附近的波数误差应不大于±5cm-1,在1000cm-1附近的波数误差应不大于±lcm-1。
红外分光光度法1 简述化合物受红外辐射照射后,使分子的振动和转动运动由较低能级向较高能及跃迁,从而导致对特定频率红外辐射的选择性吸收,形成特征性很强的红外吸收光谱,红外光谱又称振-转光谱。
红外光谱是鉴别物质和分析物质化学结构的有效手段,已被广泛应用于物质的定性鉴别、物相分析和定量测定,并用于研究分子间和分子内部的互相作用。
习惯上,往往把红外区分为3个区域,近红外区(12800~40000cm -1,0.78~2.5μ m),中红外区(4000~400cm -1 ,2.5~25 μ m)和远红外区(400~10cm -1 ,25~1000μ m)。
其中中红外区是药物分析中最常用的区域。
红外吸收与物质的关系在一定范围内服从朗伯-比尔定律,因而它也是红外分光光度法定量的基础。
红外分光光度计分为色散型和傅里叶变换型两种。
前者主要由光源、单色器(通常为光栅)、样品室、检测器、记录仪、控制盒数据处理系统组成。
以光栅为色散元件的红外分光光度计,以波数为线性刻度,以棱镜为色散元件的仪器,以波长为线性刻度。
波数与波长的换算关系如下:10000波数(cm -1)= )波长(μm傅里叶变换型红外光谱仪(简称FT-IR)则由光学台(包括光源、干涉仪、样品室和检测器)、记录装置和数据处理系统组成,由干涉图变为红外光谱需经快速傅里叶变换。
该型号仪器现已成为最常用的仪器。
2 红外分光光度计的检定所用仪器应按现行国家质量与核查技术监督局“色散型红外分光光度计鉴定规程”、“傅里叶变换红外光谱仪鉴定规程”和《中国药典》附录规定,并参考仪器说明书,对仪器定期进行校正检定。
2.1 波数准确度2.1.1 波数准确度的允差范围傅里叶变换红外光谱仪在3000cm -1附近的波数误差应不大于±5cm -1,在1000cm -1附近的波数误差应不大于±1cm -1。
2.1.2 波数准确度检定方法2.1.2.1 以聚苯乙烯膜校正按仪器使用说明书要求设置参数,以常用的扫描速度记录厚度为50μ m 的聚苯乙烯膜红外光谱图。
红外分光光度法
红外分光光度法是当物质分子吸收-记波长的光能,能引起分子振动和转动能级跃迁,产生的吸收光谱一般在2.5~25nm的中红外光区,称为红外分子吸收光谱,简称红外光谱。
利用红外光谱对物质进行定性分析或定量测定的方法称红外分光光度法。
由于物质分子发生振动和转动能级跃迁所需的能量较低,几乎所有的有机化合物在红外光区均有吸收。
分子中不同官能团,在发生振动和转动能级跃迁时所需的能量各不相同,产生的吸收谱带其波长位置就成为鉴定分子中官能团特征的依据,其吸收强度则是定量检测的依据。
红外分光光度法可用于分子结构的基础研究(测定分子键长、键角、推断分子的立体构型等),以及化学组成的分析(化合物的定性定量分析),应用最广泛的是对未知毒物的结构分析、纯度鉴定。
缺点是灵敏度低,不宜进行微量成分定量测定,而1L要求样品必须纯化。
后来发展起来的傅立叶红外光谱法克服了灵敏度低的不足,可测定(T9g的微量样品。
中文名称:红外分光光度法
英文名称:infrared spectrophotometry
定义:通过测定物质在波长2.5~25 μm(按波数计为4000~400 cm-1)的红
外光区范围内光的吸收度,对物质进行定性和定量分析的方法。
所用仪器为
红外分光光度计
仪器:红外分光光度计
流程:光源->吸收池->单色器->检测器->记录装置
分为色散型(已淘汰)和干涉型。
色散型:
光源:一般常见的为硅碳棒,特殊线圈,能斯特灯(已淘汰)。
色散元件:反射光栅
检测器:真空热电偶及Golay池
吸收池:液体池和气体池(具有岩盐窗片)
干涉型:
光源:同色散型
单色器:迈克尔逊干涉仪
检测器:多用热电性硫酸三甘肽(TGS)或光电导性检测器。
图解析
解析原则:四先四后相关法
先特征(区),后指纹(1250/cm)。
先最强(峰),后次强(峰)。
先粗查,后细找。
先否定,后肯定。
红外识谱歌
外可分远中近,中红特征指纹区,
1300来分界,注意横轴划分异。
看图要知红外仪,弄清物态液固气。
样品来源制样法,物化性能多联系。
识图先学饱和烃,三千以下看峰形。
2960、2870是甲基,2930、2850亚甲峰。
1470碳氢弯,1380甲基显。
二个甲基同一碳,1380分二半。
面内摇摆720,长链亚甲亦可辨。
烯氢伸展过三千,排除倍频和卤烷。
末端烯烃此峰强,只有一氢不明显。
化合物,又键偏,~1650会出现。
烯氢面外易变形,1000以下有强峰。
910端基氢,再有一氢990。
顺式二氢690,反式移至970;
单氢出峰820,干扰顺式难确定。
炔氢伸展三千三,峰强很大峰形尖。
三键伸展二千二,炔氢摇摆六百八。
芳烃呼吸很特征,1600~1430。
1650~2000,取代方式区分明。
900~650,面外弯曲定芳氢。
五氢吸收有两峰,700和750;
四氢只有750,二氢相邻830;
间二取代出三峰,700、780,880处孤立氢醇酚羟基易缔合,三千三处有强峰。
C-O伸展吸收大,伯仲叔醇位不同。
1050伯醇显,1100乃是仲,
1150叔醇在,1230才是酚。
1110醚链伸,注意排除酯酸醇。
若与π键紧相连,二个吸收要看准,
1050对称峰,1250反对称。
苯环若有甲氧基,碳氢伸展2820。
次甲基二氧连苯环,930处有强峰,
环氧乙烷有三峰,1260环振动,
九百上下反对称,八百左右最特征。
缩醛酮,特殊醚,1110非缩酮。
酸酐也有C-O键,开链环酐有区别,
开链强宽一千一,环酐移至1250。
羰基伸展一千七,2720定醛基。
吸电效应波数高,共轭则向低频移。
张力促使振动快,环外双键可类比。
二千五到三千三,羧酸氢键峰形宽,
920,钝峰显,羧基可定二聚酸、
酸酐千八来偶合,双峰60严相隔,
链状酸酐高频强,环状酸酐高频弱。
羧酸盐,偶合生,羰基伸缩出双峰,
1600反对称,1400对称峰。
1740酯羰基,何酸可看碳氧展。
1180甲酸酯,1190是丙酸,
1220乙酸酯,1250芳香酸。
1600兔耳峰,常为邻苯二甲酸。
氮氢伸展三千四,每氢一峰很分明。
羰基伸展酰胺I,1660有强峰;
N-H变形酰胺II,1600分伯仲。
伯胺频高易重叠,仲酰固态1550;
碳氮伸展酰胺III,1400强峰显。
胺尖常有干扰见,N-H伸展三千三,
叔胺无峰仲胺单,伯胺双峰小而尖。
1600碳氢弯,芳香仲胺千五偏。
八百左右面内摇,确定最好变成盐。
伸展弯曲互靠近,伯胺盐三千强峰宽,
仲胺盐、叔胺盐,2700上下可分辨,
亚胺盐,更可怜,2000左右才可见。
硝基伸缩吸收大,相连基团可弄清。
1350、1500,分为对称反对称。
氨基酸,成内盐,3100~2100峰形宽。
1600、1400酸根展,1630、1510碳氢弯。
盐酸盐,羧基显,钠盐蛋白三千三。
矿物组成杂而乱,振动光谱远红端。
钝盐类,较简单,吸收峰,少而宽。
注意羟基水和铵,先记几种普通盐。
1100是硫酸根,1380硝酸盐,
1450碳酸根,一千左右看磷酸。
硅酸盐,一峰宽,1000真壮观。
勤学苦练多实践,红外识谱不算难。
外分光光度法
发布日期:[2010-12-10] 共阅[784]次
附录ⅣC红外分光光度法
仪器及其校正可使用傅里叶变换红外光谱仪或色散型红外分光光度计。
用聚苯乙烯薄膜(厚度约为0.04mm)校正仪器,绘制其光谱图,用3027cm -1,2851cm -1,1601cm -1,
1028cm -1,907cm -1处的吸收峰对仪器的波数进行校正。
傅里叶变换红外光谱仪在
3000cm -1附近的波数误差应不大于±5cm -1,在1000cm -1附近的波数误差应不大于±1cm -1。
用聚苯乙烯薄膜校正时,仪器的分辨率要求在3110~2850cm -1范围内应能清晰地分辨出7个峰,峰2851cm -1与谷2870cm -1之间的分辨深度不小于18%透光率,峰
1583cm -1与谷1589cm -1之间的分辨深度不小于12%透光率。
仪器的标称分辨率,除另
有规定外,应不低于2cm -1。
供试品的制备及测定
1.原料药鉴别除另有规定外,应按照国家药典委员会编订的《药品红外光谱集》各卷收载的各光谱图所规定的方法制备样品。
具体操作技术参见《药品红外光谱集》的
说明。
采用固体制样技术时,最常碰到的问题是多晶现象,固体样品的晶型不同,其红外
光谱往往也会产生差异。
当供试品的实测光谱与《药品红外光谱集》所收载的标准光谱
不一致时,在排除各种可能影响光谱的外在或人为因素后,应按该药品光谱图中备注的
方法或各品种正文中规定的方法进行预处理,再绘制光谱,比对。
如未规定该品供药用
的晶型或预处理方法,则可使用对照品,并采用适当的溶剂对供试品与对照品在相同的
条件下同时进行重结晶,然后依法绘制光谱,比对。
如已规定特定的药用晶型,则应采
用相应晶型的对照品依法比对。
当采用固体制样技术不能满足鉴别需要时,可改用溶液法测定光谱后比对。
2. 制剂鉴别品种鉴别项下应明确规定制剂的前处理方法,通常采用溶剂提取法。
提取时应选择适宜的溶剂,以尽可能减少辅料的干扰,并力求避免导致可能的晶型转变。
提取的样品再经适当干燥后依法进行红外光谱鉴别。
3. 多组分原料药鉴别不能采用全光谱比对,可借鉴注意事项(3)的方法,选择
主要成分的若干个特征谱带,用于组成相对稳定的多组分原料药的鉴别。
1
4.晶型、异构体限度检查或含量测定供试品制备和具体测定方法均按各品种项下有关规定操作。
注意事项
1.各品种项下规定"应与对照的图谱(光谱集××图)一致",系指《药品红外光
谱集》第一卷(1995年版)、第二卷(2000年版)和第三卷(2005年版)的图谱。
同
一化合物的图谱若在不同卷上均有收载时,则以后卷所收的图谱为准。
2.药物制剂经提取处理并依法录制光谱,比对时存在如下四种可能:
(1)辅料无干扰,待测成分的晶型不变化,此时可直接与原料药的标准光谱进行
比对;
( 2)辅料无干扰,但待测成分的晶型有变化,此种情况可用对照品经同法处理后
的光谱比对;
( 3)待测成分的晶型不变化,而辅料存在不同程度的干扰,此时可参照原料药的
标准光谱,在指纹区内选择3~5个不受辅料干扰的待测成分的特征谱带,以这些谱带的位置(波数值)作为鉴别的依据。
鉴别时,实测谱带的波数误差应小于规定值的0.5%;
( 4)待测成分的晶型有变化,辅料也存在干扰,此种情况使问题变得复杂,故一
般不宜采用红外鉴别。
3.由于各种型号的仪器性能不同,供试品制备时研磨程度的差异或吸水程度不同等原因,均会影响光谱的形状。
因此,进行光谱比对时,应考虑各种因素可能造成的影响。