结合案例谈医疗器械稽查方法
- 格式:ppt
- 大小:788.00 KB
- 文档页数:37
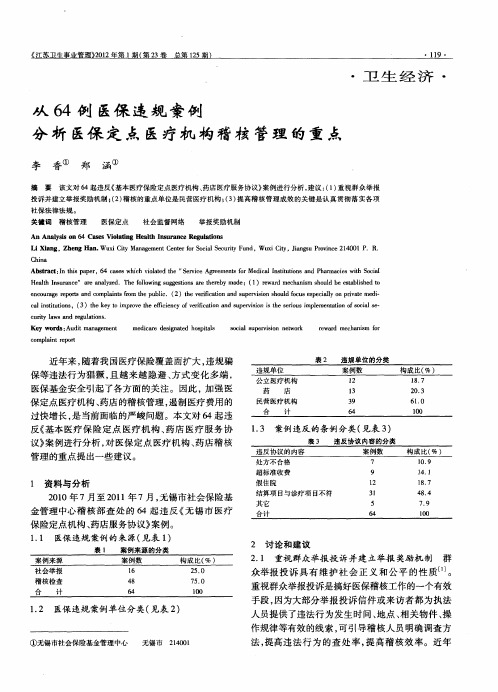

骨科植入性医疗器械常见违法现象及监管对策骨科植入性医疗器械在医疗器械分类中属Ⅲ类,是植入人体的高风险医疗器械,其安全性、有效性必须严格控制并要求得到充分保障。
它具有三个特点:第一,可追溯性。
2003年3月18日,国家药品监管局和卫生部联合印发的《关于继续加强对医疗机构的医疗器械监督管理的通知》中要求,各级各类医疗机构对骨科植入性医疗器械必须建立详细的使用记录,记录至少应包括患者姓名、产品名称、产品数量、规格型号等必要的产品跟踪信息,使产品具有可追溯性;第二,间接性。
植入人体后,监管过程不能直接看到其实物;其三,回收性。
使用一定时间后必须从人体取出。
鉴于该类医疗器械的高风险特点,笔者对骨科植入性医疗器械常见违法现象进行了梳理,并结合实际案例,谈谈骨科植入性医疗器械的监管。
违法现象一:擅自改变规格型号骨科植入性医疗器械规格型号分类较细,品种繁多,那些分类细、型号多的产品规格型号常用附件来登载。
改变规格型号大致分为两种情况,其一,规格型号代号不变,改变与代号相对应的骨科植入器械的具体尺寸;其二,完全改变规格型号,器械上所标示的规格型号与注册证上的完全不符。
如某局在监督中发现,某医院使用的金属股骨颈固定钉产品注册证书附件《医疗器械产品生产制造认可表》规格型号标明“GKSD:L 85~120mm GWPD:70-120mm”,合格证明上标示规格型号“双头空心80mm”。
GSKD即空心双头加压,在注册证上已限定它的规格为85-120mm长,而这颗金属股骨颈固定钉长度却为80mm,不在批准范围。
监管对策:医疗器械注册证附件《医疗器械产品注册登记表》(进口医疗器械)或《医疗器械产品制造认可表》(国产医疗器械)会有一栏“规格型号”对所批准的医疗器械型号进行限定。
将合格证明上所标示的规格型号与产品注册证书附件上的规格型号进行对比,如果发现有出入,应当深入调查取证,明确规格型号的改变是文字上的改变还是实质上的改变。
如果只是文字上的改变,只需要求生产企业及时变更产品注册证书即可;如果是产品本身发生改变,就应当按无产品注册证书进行处罚。

医疗器械不良事件监测及风险控制管理方法医疗器械不良事件的监测及风险控制管理方法随着科技的不断发展,医疗器械在医疗领域的应用越来越广泛。
然而,医疗器械不良事件也随之增加,给患者的健康带来了一定的风险。
因此,监测医疗器械不良事件并进行风险控制管理显得尤为重要。
本文将介绍医疗器械不良事件监测的方法以及风险控制管理的策略。
一、医疗器械不良事件监测的方法1.主动监测:医疗机构应建立健全医疗器械不良事件的主动监测机制。
通过设立不良事件报告系统,规范医务人员对不良事件的报告和记录,及时发现和上报医疗器械不良事件。
同时,医疗机构可以建立与药监部门的信息沟通渠道,及时了解国内外医疗器械不良事件的动态。
2.被动监测:药监部门可以通过建立医疗器械不良事件报告平台,接受医务人员和患者的不良事件报告。
同时,可以通过定期抽样检查、定期回访等方式,了解医疗器械在使用过程中可能存在的问题。
3.数据分析:通过对医疗器械不良事件的数据进行分析,可以及时识别存在的问题和风险。
药监部门可以利用数据分析的结果,对医疗器械进行分类管理,针对性地制定监管措施和政策。
二、医疗器械不良事件的风险控制管理策略1.风险评估:对医疗器械进行风险评估,确定其安全性和可靠性。
通过对器械的设计、材料、工艺等方面进行评估,减少不良事件的发生。
2.质量控制:医疗器械生产企业应建立完善的质量控制体系,严格按照相关法规和标准进行生产制造。
同时,医疗机构应加强对医疗器械的采购和验收,确保所购买的器械符合质量要求。
3.信息共享:加强医疗器械不良事件的信息共享,提高医务人员和患者的风险意识。
通过建立信息平台,及时向医务人员和患者发布医疗器械不良事件的警示信息,提醒他们注意使用医疗器械的安全性和可靠性。
4.培训教育:加强医务人员的培训教育,提高其对医疗器械的正确使用和操作能力。
医疗机构可以定期组织医务人员的培训,加强对医疗器械不良事件的案例分析和经验总结,提高其对不良事件的识别和处理能力。

医疗器械违规操作案例分析在医疗领域,医疗器械的正确使用对于患者的诊断、治疗和康复至关重要。
然而,不幸的是,我们时常会听到一些关于医疗器械违规操作的案例,这些案例不仅给患者的健康带来了潜在风险,也严重影响了医疗行业的信誉和形象。
本文将对一些典型的医疗器械违规操作案例进行深入分析,以期引起大家对这一问题的重视,并从中汲取教训。
案例一:某医院使用未经注册的医疗器械在具体城市的一家医院,被发现使用了一批未经注册的医疗器械。
这些器械包括某种新型的心脏监测设备和微创手术工具。
医院为了追求所谓的“先进技术”,从非法渠道引进了这些没有经过国家相关部门严格审批和注册的产品。
这种违规操作带来了诸多严重后果。
首先,未经注册的医疗器械其安全性和有效性无法得到保障。
这些器械可能没有经过充分的临床试验和质量检测,在使用过程中容易出现故障或不准确的测量结果,从而影响医生对患者病情的判断和治疗决策。
其次,使用未注册的器械违反了国家的法律法规,医院面临着严厉的处罚,包括罚款、停业整顿等。
这不仅损害了医院的声誉,也给患者带来了极大的不安和不信任感。
造成这一违规行为的原因是多方面的。
一方面,医院管理层存在急功近利的心态,为了提升医院的竞争力和知名度,盲目追求新技术,而忽视了合法合规的重要性。
另一方面,相关监管部门的监管力度不够,未能及时发现和制止这种非法行为。
案例二:医疗器械操作人员未经专业培训在另一个案例中,具体城市的一家小型诊所被曝光其医疗器械操作人员未经专业培训就上岗操作。
例如,负责操作 B 型超声诊断仪的工作人员,仅仅通过简单的自学和观看操作视频,就开始为患者进行检查。
这种情况导致了诊断结果的不准确和不可靠。
B 型超声诊断需要操作人员具备扎实的医学知识和丰富的实践经验,才能准确地解读图像并做出正确的诊断。
未经专业培训的操作人员可能会误判病情,给患者带来误诊和延误治疗的风险。
究其原因,主要是诊所为了降低成本,没有安排员工参加正规的培训课程,也没有聘请有资质的专业人员。

医疗器械合规审核案例分析概述:医疗器械是保障人类健康的重要工具,然而,市场上的许多医疗器械存在合规问题,因此对医疗器械的合规性进行审核十分必要。
本文将以具体案例分析的方式,探讨医疗器械合规审核的重要性以及过程中的挑战和解决方案。
案例1:患者使用非合格医疗器械的后果某医院在购买了一种新型阿司匹林自动注射器后,未进行合规审核就将其投入使用。
然而,该器械未通过国家药监部门的认证,并存在输送精准剂量不准确的问题。
因此,一名患者在注射时受到过量药物的影响,导致身体不适。
医院被迫停止使用该器械,并承担赔偿责任。
这个案例显示了医院未进行合规审核带来的直接后果。
医疗器械的合规审核是确保器械性能和安全性的重要步骤。
医院在购买和使用医疗器械之前,应该仔细审查器械的合规性,确保其符合国家和行业准则。
案例2:医疗器械企业合规审核的实践某医疗器械企业开发了一种新型体外液体处理器。
为了确保其合规性,企业决定进行合规审核。
审核团队根据国家和地区的法规要求,对产品性能、材料、生产工艺等方面进行全面检查和评估。
在审核过程中,团队发现了一些潜在的合规问题,如使用的材料可能对患者产生过敏反应。
企业随即调整材料选择,并重新测试和验证产品的合规性。
最终,经过持续努力,该企业成功使产品符合国家和地区的合规要求,并获得了销售许可证。
这个案例突显了医疗器械企业合规审核的重要性和挑战。
企业应积极主动地进行合规审核,确保产品符合法规要求。
合规审核团队的专业知识和细致的工作是确保产品合规性的关键。
同时,企业还需要灵活应对可能出现的合规问题,采取有效措施解决并确保产品的合规性。
解决方案:合规审核的有效实施针对医疗器械的合规审核,可以采取以下几个重要步骤:1.确立合规标准:明确国家和地区的法规要求以及行业标准,以及市场准入的相关规定。
2.审核人员培训:组建合规审核团队,并对团队成员进行培训,提高其对合规审核的理解和能力。
3.产品现场检查:对生产现场、设备、材料等进行全面检查,确保无违规操作和不合规物质的使用。
