苯基WHE
- 格式:pdf
- 大小:25.91 KB
- 文档页数:4

关于苯的详细资料。
苯(benzene,C6H6),有机化合物,是组成结构最简单的芳香烃,在常温下为一种无色、有甜味的透明液体,并具有强烈的芳香气味。
苯可燃,有毒,为IARC一类致癌物。
苯难溶于水,易溶于有机溶剂,本身也可作为有机溶剂。
苯是一种石油化工基本原料。
苯的产量和生产的技术水平是一个国家石油化工发展水平的标志之一。
苯具有的环系叫苯环,是最简单的芳环。
苯分子去掉一个氢以后的结构苯与苯基叫苯基,用Ph表示。
因此苯也可表示为PhH。
CAS号: 71-43-2RTECS号: CY1400000SMILES: C1=CC=CC=C1化学式: C6H6摩尔质量: 78.11 g /mol密度: 0.8786 g/mL熔点: 278.65 K (5.5 ℃)沸点: 353.25 K (80.1 ℃)在水中的溶解度: 0.18 g/ 100 ml 水标准摩尔熵So298: 173.26 J/mol·K标准摩尔热容 Cpo: 135.69 J/mol·K (298.15 K)闪点: -10.11℃(闭杯)自燃温度: 562.22℃1.发现历史苯最早是在18世纪初研究将煤气作为照明用气时合成出来的。
1803年~1819年G. T. Accum制出了许多产品,其中一些样品用现代的分析方法检测出有少量的苯。
1825年,迈克尔·法拉第(Michael Faraday)从鱼油等类似物质的热裂解产品中分离出了较高纯度的苯,称之为“氢的重碳化物”(Bicarburet of hydrogen)。
并且测定了苯的一些物理性质和它的化学组成,阐述了苯分子的碳氢比。
1833年,Milscherlich确定了苯分子中6个碳和6个氢原子的实验式(C6H6)。
1845年德国化学家霍夫曼从煤焦油的轻馏分中发现了苯,他的学生C. Mansfield随后进行了加工提纯。
后来他又发明了结晶法精制苯。
他还进行工业应用的研究,开创了苯的加工利用途径。

【了解职业危害】系列之——苯及其化合物苯(C6H6)是一种有机化合物。
在常温下,苯是一种无色、有甜味的透明液体,并具有强烈的芳香气味。
进入工业时代以来,苯成为一种非常重要的化工原料。
我们日常生活中的很多用品都离不开苯及其化合物:①苯与乙烯生成乙苯,后者可以用来生产制塑料的苯乙烯;②苯与丙烯生成异丙苯,后者可以经异丙苯法来生产丙酮与制树脂和粘合剂的苯酚;制尼龙的环己烷;③合成顺丁烯二酸酐,用于制造聚酯树脂、醇酸树脂、农药、富马酸、纸张处理剂等。
④用于制作苯胺的硝基苯;苯胺主要用于医药和橡胶硫化促进剂,也是制造树脂和涂料的原料。
⑤用于农药的各种氯苯;⑥合成用于生产洗涤剂和添加剂的各种烷基苯。
⑦合成氢醌,蒽醌等化工产品。
…………可怕的苯中毒:血癌杀手苯是一种典型的双刃剑,极大的丰富便利我们生活的同时,也埋伏下安全隐患的种子。
苯被IARC(国际癌症研究机构)列为一类致癌物,其化合物也多有毒性,呼吸、接触都可能发生苯中毒,因而苯中毒已经成为最严重的职业危害之一。
苯中毒的案例很多,例如:河北省白沟镇箱包加工企业发生苯中毒致6人死亡;浙江温岭制鞋企业发生正己烷中毒4人死亡;北京某包装厂发生20人苯中毒致1人死亡等。
急性苯中毒苯中毒轻微者酒醉状,伴恶心、呕吐、步态不稳、幻觉、哭笑失常等,表现重者意识丧失、肌肉痉挛、或抽搐、血压下降、瞳孔散大、个别病例可有心室颤动,可因呼吸麻痹死亡。
慢性苯中毒慢性苯中毒除影响神经系统外,还影响造血系统,导致再生障碍性贫血,甚至白血病……苯中毒是由什么原因引起的?苯是现代工业社会不可缺少的重要的化学原料之一,是蜡、树脂、油的溶剂,合成化学制品和制药的中间体,86%苯用于制造苯乙烯、苯酚、环乙烷和其他有机物。
剩余部分主要用于制造洗涤剂、杀虫剂和油漆清除剂。
苯可作为汽油一种成份,含量<2% 。
苯可以以蒸气的形态经呼吸道吸收,或者以液体形态经消化道吸收完全。
皮肤也可以吸收少量苯。
苯以蒸气状态经呼吸道吸入,进入体内后,部分以原形由肺呼出;部分在肝脏代谢,通过微粒体混合功能氧化酶进行羟化,转化为酚、对苯二酚、邻苯二酚等酚类代谢产物。

苯苯苯(Benzene, C6H6)⼀种碳氢化合物即最简单的芳烃,在常温下是甜味、可燃、有致癌毒性的⽆⾊透明液体,并带有强烈的芳⾹⽓味。
它难溶于⽔,易溶于有机溶剂,本⾝也可作为有机溶剂。
苯具有的环系叫苯环,苯环去掉⼀个氢原⼦以后的结构叫苯基,⽤Ph表⽰,因此苯的化学式也可写作PhH。
苯是⼀种⽯油化⼯基本原料,其产量和⽣产的技术⽔平是⼀个国家⽯油化⼯发展⽔平的标志之⼀。
2017年10⽉27⽇,世界卫⽣组织国际癌症研究机构公布的致癌物清单初步整理参考,苯在⼀类致癌物清单中。
[1]中⽂名苯英⽂名Benzene,benzol,benzeen别称安息油化学式C6H6分⼦量78.11CAS登录号71-43-2EINECS登录号200-753-7熔点5.5ºC沸点80ºC⽔溶性0.18g/100ml密度0.8765(20) g/cm3闪点-11ºC应⽤⽤作⾹料、染料、塑料、医药、炸药、橡胶等危险性描述易燃物质,有毒物质危险品运输编号UN1114/1115化学品类别有机物--苯的同系物更多发现历史凯库勒双键摆动模型苯是在1825年由英国科学家法拉第(Michael Faraday,1791— 1867)⾸先发现的。
19世纪初,英国和其他欧洲国家⼀样,城市的照明已普遍使⽤煤⽓。
从⽣产煤⽓的原料中制备出煤⽓之后,剩下⼀种油状的液体却长期⽆⼈问津。
法拉第是第⼀位对这种油状液体感兴趣的科学家。
他⽤蒸馏的⽅法将这种油状液体进⾏分离,得到另⼀种液体,实际上就是苯。
当时法拉第将这种液体称为“氢的重碳化合物”。
1834年,德国科学家⽶希尔⾥希(E·E·Mitscherlich,1794— 1863)通过蒸馏苯甲酸和⽯灰的混合物,得到了与法拉第所制液体相同的⼀种液体,并命名为苯。
待有机化学中的正确的分⼦概念和原⼦价概念建⽴之后,法国化学家⽇拉尔(C·F·Gerhardt,1815— 1856)等⼈⼜确定了苯的相对分⼦质量为78,分⼦式为C6H6。

n-苯基-1,4-苯二胺是一种有机化合物,化学式为C12H11N。
它是苯胺的衍生物,具有两个苯环和一个间苯二胺基团。
它是一种固体,常用于有机合成反应中。
1. 基本结构n-苯基-1,4-苯二胺分子由一个苯环和一个1,4-苯二胺基团构成。
苯环由六个碳原子和五个氢原子组成,1,4-苯二胺基团由一个苯环和两个氨基组成。
两个苯环通过1,4-位置连接在一起,构成了n-苯基-1,4-苯二胺的基本结构。
2. 物理性质n-苯基-1,4-苯二胺是一种白色结晶固体,具有良好的溶解性和热稳定性。
它可以溶解在许多有机溶剂中,如乙醇、丙酮和甲醇等。
在热稳定性方面,n-苯基-1,4-苯二胺在高温下不会分解或失去活性,因此在有机合成反应中具有一定的优势。
3. 化学性质n-苯基-1,4-苯二胺具有一定的化学反应活性,可以与许多化合物发生反应,如醛、酮、羧酸和酰氯等。
在有机合成反应中,它常用作中间体或催化剂,参与芳香族化合物的合成和官能团的引入等反应。
4. 应用领域n-苯基-1,4-苯二胺在有机合成领域有着广泛的应用。
它常用于染料、药物、高分子材料和化学品的合成中。
在染料合成中,n-苯基-1,4-苯二胺可以作为重要的中间体,参与染料分子的构建和着色反应。
在药物合成中,它可以作为药物分子的骨架或功能基团,影响药物的性质和活性。
在高分子材料和化学品的合成中,它可以参与聚合反应或官能团的引入,影响材料的性能和用途。
5. 安全性n-苯基-1,4-苯二胺在使用和储存过程中需要注意其安全性。
它具有一定的毒性和刺激性,对皮肤和呼吸道有一定的危害。
因此在操作时需要采取相应的防护措施,如佩戴防护手套和口罩等。
需要注意其在储存和运输过程中避免受潮和高温等因素的影响,以保证其质量和稳定性。
n-苯基-1,4-苯二胺是一种重要的有机化合物,具有良好的溶解性、热稳定性和化学反应活性,广泛应用于染料、药物、高分子材料和化学品的合成中。
在使用时需要注意其安全性,合理选择和操作,以保证其在合成过程中的效果和安全性。

化学品安全技术说明书苯基硫醇第一部分化学品及企业标识化学品中文名:苯基硫醇化学品英文名:phenyl mercaptan;thiophenol供应商名称:****化工有限公司供应商名称:天津****化工有限公司供应商地址:天津市**区**路**号**室供应商电话:4571-5858****邮编:248***供应商传真:4571-5858****电子邮件地址:4527**************产品推荐及限制用途:不溶于水,可混溶于乙醇、乙醚、苯、二硫化碳。
第二部分危险性概述紧急情况概述:易燃液体和蒸气,吞咽致命,皮肤接触会致命,吸入致命。
GHS危险性类别:易燃液体-类别3;急性毒性-经口-类别2;急性毒性-经皮-类别2;急性毒性-吸入-类别1;皮肤腐蚀/刺激-类别2;严重眼损伤/眼刺激-类别2A;生殖毒性-类别2;特异性靶器官毒性-一次接触-类别2;特异性靶器官毒性-一次接触-类别3(呼吸道刺激);特异性靶器官毒性-反复接触-类别1;危害水生环境-急性危害-类别1;危害水生环境-长期危害-类别1标签要素:象形图:警示词:危险危险信息:H226:易燃液体和蒸气H300:吞咽致命H310:皮肤接触会致命H330:吸入致命H315:造成皮肤刺激H319:造成严重眼刺激H361:怀疑对生育能力或胎儿造成伤害(如果已知,说明特异性效应;如果确证无其他接触途径引起危害,说明接触途径)H371:可能对器官造成损害(如果知道,说明所受损的器官)(如果可确证无其他接触途径引起危害,说明接触途径)H335:可能引起呼吸道刺激H372:长时间或反复接触(如果可确证无其他接触途径引起该危害,说明接触途径)对器官造成伤害(如果已经知道,说明所受损害的器官)H410:对水生生物毒性极大并具有长期持续影响防范说明:预防措施:P210:远离热源/火花/明火/热表面。
禁止吸烟。
P233:保持容器密闭。
P240:容器和接收设备接地/等势联接。

第一部分化学品及企业标识化学品中文名:苯基硫醇化学品英文名:phenyl mercaptan化学品别名:苯硫酚;巯基苯;硫代苯酚CAS No.:108-98-5EC No.:203-635-3分子式:C6H6S分子量:110.18产品推荐用途:主要用于农药、医药的合成。
第二部分危险性概述紧急情况概述:液体。
易燃,其蒸气与空气混合,能形成爆炸性混合物。
吞食后有剧毒。
跟皮肤接触有剧毒。
对皮肤有刺激性。
对眼睛有严重刺激性。
吸入有剧毒。
对呼吸道有刺激作用。
可能有损伤胎儿或胚胎的危险。
短期暴露有损伤健康的危险。
长期暴露有严重损伤健康的危险。
对水生物有剧毒,使用适当的容器,以预防污染环境。
对水生环境可能会引起长期有害作用。
使用适当的容器,以预防污染环境。
GHS危险性类别:根据GB30000-2013化学品分类和标签规范系列标准(参阅第十六部分),该产品分类如下:易燃液体,类别3;急毒性-口服,类别2;急毒性-皮肤,类别2;皮肤腐蚀/刺激,类别2;眼损伤/眼刺激,类别2A;急毒性-吸入,类别1;特定目标器官毒性-单次接触:呼吸道刺激,类别3;生殖毒性,类别2;特定目标器官毒性-单次接触,类别2;特定目标器官毒性-重复接触,类别1;危害水生环境-急性毒性,类别1;危害水生环境-慢性毒性,类别1。
标签要素象形图:警示词:危险危险信息:易燃液体和蒸气,吞咽致命,皮肤接触致命,造成皮肤刺激,造成严重眼刺激,吸入致命,可能造成呼吸道刺激,怀疑对生育能力或胎儿造成伤害,可能对器官造成损害,长期或重复接触会对器官造成伤害,对水生生物毒性极大,并具有长期持续影响。
预防措施:使用前取得专业说明。
在阅读并明了所有安全措施前切勿搬动。
远离热源、热表面、火花、明火以及其它点火源。
禁止吸烟。
保持容器密闭。
容器和接收设备接地和等势联接。
使用不产生火花的工具。
采取措施,防止静电放电。
不要吸入粉尘/烟/气体/烟雾/蒸气/喷雾。
严防进入眼中、接触皮肤或衣服。


苯基丁二酸用途
苯基丁二酸,又称为1,4-丁二酸苯基酯,是一种有机化合物。
它可以作为一种重要的化学品,具有广泛的用途。
首先,苯基丁二酸是一种重要的合成原料,可以用于制备聚酯类高分子材料。
例如,在纤维、管道等领域中常用的聚酯树脂就可以通过苯基丁二酸和乙二醇的缩合反应制备而成。
其次,苯基丁二酸也被广泛用于生产涂料和塑料。
在涂料领域中,它可以作为涂料中的主要成分,提高涂料的耐久性和抗腐蚀性。
在塑料领域中,苯基丁二酸可以作为增塑剂,增加塑料的柔软性、韧性和延展性。
此外,苯基丁二酸还可以用于生产医药、染料、香料和化妆品等产品。
在医药领域中,它可以作为制备某些药物的中间体;在染料和香料领域中,它则可以作为染料和香料的原料;在化妆品领域中,苯基丁二酸可以用于制作某些护肤品和化妆品的成分。
总之,苯基丁二酸是一种重要的有机化合物,可以用于制备聚酯、涂料、塑料、医药、染料、香料和化妆品等产品。
由于其广泛的用途和应用,苯基丁二酸在各个行业中都有着重要的地位。
- 1 -。

苯酚四苯基磷闪点-回复"苯酚四苯基磷闪点"是一个化学领域内的专有名词,指的是具有苯基作为取代基的苯酚四苯基磷化合物的闪点。
为了解释这个专有名词,我们需要先了解一些相关的基本背景知识,然后从苯酚、四苯基磷以及闪点的定义和原理等方面逐步解释。
以下是一步一步回答的详细解释:一、苯酚:苯酚是一种有机化合物,也被称为羟基苯,化学式为C6H6O。
它是苯环上连接一个羟基(OH基)的化合物,常见的化学名字是苯酚。
苯酚具有毒性、易燃和腐蚀性,在工业和实验室中有广泛应用。
二、四苯基磷:四苯基磷是一种有机磷化合物,化学式为P(C6H5)4。
它是一个磷原子与四个苯基连接而成的化合物。
四苯基磷是一种常用的有机磷反应试剂,广泛应用于有机合成领域。
三、闪点:闪点是液体沸腾状态下的最低温度,也就是液体在给定条件下开始产生可燃气体的温度。
当液体的闪点达到或超过环境温度时,会发生闪燃或爆炸。
闪点是评估液体易燃性和危险性的重要参数。
根据以上基本背景知识,我们可以更全面地解释“苯酚四苯基磷闪点”。
苯酚四苯基磷是将苯酚和四苯基磷进行化合反应得到的化合物,其化学式为C6H6O-P(C6H5)4。
由于四苯基磷是一种有机磷反应试剂,在该化合物中起到了类似于取代基的作用。
苯酚四苯基磷具有独特的化学性质和应用领域。
闪点是评估液体易燃性和危险性的参数之一。
苯酚四苯基磷的闪点是指在给定条件下,该化合物开始产生可燃气体的最低温度。
详细了解苯酚四苯基磷闪点的性质和应用需要一定的专业化知识。
首先,我们需要知道该化合物的化学性质,包括其分子结构、化学键和各个基团的性质等。
这些信息可以通过实验数据和文献资料获得。
然后,我们需要了解闪点的原理和测量方法。
闪点的测量方法通常采用闭杯法或开杯法。
闭杯法是将待测液体样品加入闭杯中,加热样品,然后用点火源靠近液体表面,观察是否发生闪燃;开杯法是将液体样品倒入开杯中,然后点火,测量液体开始闪燃的温度。
这两种方法都可以用于测量化合物的闪点。
T1——450℃<TT2——300<T≤450℃T3——200<T≤300℃T4——135<T≤200℃T5——100<T≤135T6——85<T≤100℃3、危害级别:DIPHYL DT—0(无)人体危险4——表示只要很短接触,即使立即给予医疗处理也能致死或造成严重残留伤害的物质;3——表示只要短期接触,即使立即给予医疗处理也能引起严重的暂时或残留健康危害的物质;2——在强烈或连续接触时,会引起暂时机能丧失或可能的残留伤害,除非立即给予医疗处理的物质;1——接触时引起轻微刺激,但只有很小的残留伤害,即使不给予治疗的物质;0——着火条件下接触,对人体的危害低于普通可燃物质的物质。
4、燃烧级别:DIPHYL DT——G1易燃性4——指在大气压力和正常环境温度下,迅速或完全气化的物质,或者容易在空气中扩散并容易燃烧的物质;3——在几乎所有的环境温度下,都能着火的液体和固体;2——指在发生燃烧以前,必须适当加热或接触相当高环境温度的物质;1——必须经预热才能燃着的物质;0——指不燃物质。
5、爆炸极限:DIPHYL DT上限约14.5(按体积百分比)(206℃),下限约0.8(按体积百分比)(132℃)可燃气体、可燃液体蒸气或可燃粉尘与空气组成的混合物,并非任何混合比例下都可以爆炸,而是固定浓度范围的,不同可燃物有不同的固定浓度范围。
这一固定范围通常叫该物质的爆炸范围或爆炸极限,通常用可燃气体、可燃液体蒸气、可燃粉尘在空气中的体积百分数表示。
能够产生爆炸的最低浓度成称为爆炸下限,最高浓度为爆炸上限。
爆炸极限是在常温、常压等标准条件下测定出来的,这一范围随着温度、压力的变化而有变化。
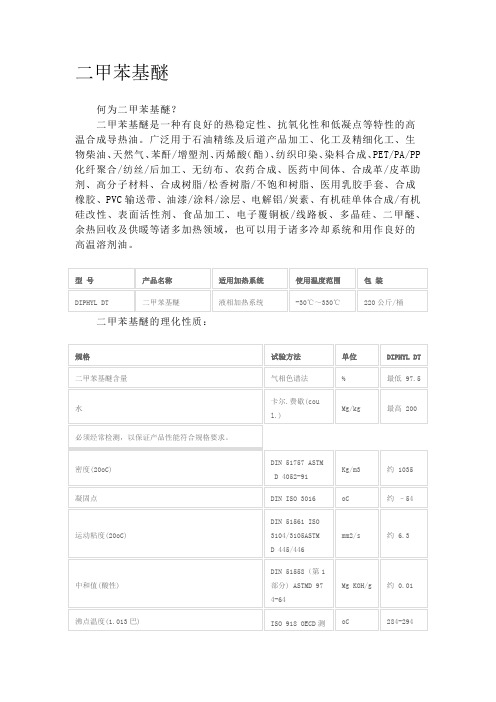
1.成分:二甲苯基醚Cas NO.:28299-41-4危害标志:无同义:二甲基二苯基醚,及其异构体混合物2.公害说明对水生有机物有毒害性,可以引起长期的水生环境不良影响。
3.急救措施轻微情况:移开污染源与皮肤接触:用肥皂涂抹后大量清水清洗。


Access to Both Anomers of Pectenotoxin Spiroketals by KineticSpiroketalizationPetri M. Pihko* and Jatta E. AhoLaboratory of Organic Chemistry, Helsinki University of Technology, P.O.B. 6100, FI-02015 HUT,FinlandPetri.Pihko@hut.fiSupporting InformationGeneral Methods. All reactions were carried out under an argon atmosphere in flame-dried glassware, unless otherwise noted. Nonaqueous reagents were transferred under argon via syringe or cannula and dried prior to use. Toluene and Et3N were distilled from Na. THF and Et2O were distilled from Na/benzophenone. CH2Cl2 and DMSO were distilled from CaH2. DMF was distilled from molecular sieves (4 Å) and oxalyl chloride was fractionally distilled. A stock sample of dry TBHP was dried by the procedure of Sharpless and co-workers.1 All batches of TBHP were also dried on 4 Å molecular sieves immediately prior to use. All of the self-made reagents were azeotropically dried with toluene. Other solvents and reagents were used as obtained from supplier, unless otherwise noted. Analytical TLC was performed using Merck silica gel F254 (230-400 mesh) plates and analyzed by UV light or by staining upon heating with vanillin solution (6 g vanillin, 5 mL conc. H2SO4, 3 mL glacial acetic acid, 250 mL EtOH) or with ninhydrin solution (1 g ninhydrin, 100 mL isopropanol, 5 drops glacial acetic acid). For silica gel chromatography, the flash chromatography technique was used, with Merck silica gel 60 (230-400 mesh) and p.a. grade solvents unless otherwise noted.The 1H NMR and 13C NMR spectra were recorded in either CDCl3 or CD3CN on a Bruker Avance 400 (1H 399.98 MHz; 13C 100.59 MHz) spectrometer. The chemical shifts are reported in ppm relative to CHCl3 (δ 7.26) or CHD2CN (δ 1.94) for 1H NMR. For the 13C NMR spectra, the residual CDCl3 (δ 77.0) or CD3CN (δ 118.26) were used as the internal standard. The enantiomeric excess (ee) of the products were determined by HPLC in comparison to the corresponding racemic samples using Waters 501 pump and Waters 486 detector, Daicel Chiralcel AD or OD columns and i-PrOH/hexane as eluent. IR spectra were recorded on a Perkin-Elmer Spectrum One spectrometer. Optical rotations were obtained with a Perkin-Elmer 343 polarimeter. High resolution mass spectrometric data were obtained by the University of Oulu on Micromass LCT spectrometer. The elemental analyses were performed at the Analytical Services of the Department of Chemical Technology, Laboratory of Organic Chemistry.The racemic samples corresponding to the compounds 18, 19, and 20 were prepared using m-CPBA (120 mol% in CH2Cl2) as the oxidant for 17. All racemic samples were purified and all subsequent reactions were performed in a manner identical to their enantioenriched counterparts.5-BenzyloxypentanolBnOTo neat 1,5-pentanediol (47.3 mL, 46.9 g, 450 mmol) at rt was added both benzyl bromide (15.3 mL, 22.1 g, 129 mmol) and powdered KOH (30.4 g, 542 mmol) in four equal portions over 1 h. After the last addition the solution was stirred for an additional 4 h. H2O (75 mL) was then added. The separated aqueous phase was extracted with EtOAc (4 x 30 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated. Silica gel column chromatography (30-70% ethyl acetate in hexanes) yielded 5-benzyloxypentanol (20.1 g, 80%) as a pale yellow oil. IR- and 1H NMR-data match those reported in literature.25-(Benzyloxy)pentanal 15BnO H15A solution of oxalyl chloride (1.9 mL, 2.74 g, 21.6 mmol) in CH2Cl2 (20 mL) was cooled to -55 ºC and DMSO (3.1 mL, 3.37mg, 43.1 mmol) was added. After 5 min, a solution of 5-benzyloxypentanol (3.81 g, 19.6 mmol) in CH2Cl2 (45 mL) was added. The resulting mixture was stirred for 15 min. Triethylamine (13.1 mL, 9.52 g, 94.1 mmol) was then added dropwise. Stirring was continued at -55 ºC for an additional 10 min and then the mixture was allowed to warm to rt. H2O (100 mL) was added and the separated aqueous phase was extracted with CH2Cl2 (2 x 60 mL). The combined organic phases were washed with brine (2 x 60 mL), dried over MgSO4, filtered and concentrated. Silica gel column chromatography (30% ethyl acetate in hexanes) yielded 5-(benzyloxy)pentanal 15 (3.5 g, 93%) as a pale yellow oil. IR- and 1H NMR-data match those reported in the literature.27-(Benzyloxy)-methyl-2-(Z)-hepteonate 16CO2MeBnO16To a solution of bis(o-cresyl) phosphonoacetate (7.4 mL, 9.03 g, 27.0 mmol) in THF (150 mL) at 0 ºC was added NaI (3.12 g, 20.8 mmol). After 5 min, NaH (60% dispersion, 0.65 g, 27 mmol) was slowly added and the resulting mixture was cooled -78 ºC. A solution of 5-(benzyloxy)pentanal 15 (4.0 g, 20.8 mmol) in THF (15 mL) was added dropwise via cannula. The resulting solution was stirred at -78 ºC for an additional 8 h before half-saturated NH4Cl (120 mL) was added. H2O (50 mL) was added to get a clear solution and the mixture was allowed to warm to rt. The separated aqueous phase was extracted with Et2O ( 3 x 80 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (5% ethyl acetate in hexanes) yielded 7-(benzyloxy)-methyl-2-(Z)-heptenoate 16 (4.39 g, 85%, Z/E 97/3) as a pale yellow oil.R f= 0.63 (50 % EtOAc in hexanes, UV/vanillin); IR (thin film, cm-1) 2944, 2859, 1723, 1645, 1438, 1199, 1170, 1103, 736, 698; 1H NMR (400 MHz, CDCl3) δ 7.37-7.25 (m, 5H), 6.22 (dt, 1H, J = 11.5 Hz, 7.5 Hz), 5.78 (dt, 1H, J = 11.5 Hz, 1.7 Hz), 4.50 (s, 2H), 3.70 (s, 3H), 3.49 (t, 2H, J = 6.4 Hz), 2.68 (dq, 2H, J = 1.7 Hz, 7.5 Hz), 1.70-1.63 (m, 2H), 1.60-1.50 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.8, 150.4, 138.6, 128.3, 127.6, 127.5, 119.4, 72.9, 70.0, 51.0, 29.3, 28.7, 25.6; HRMS (ESI) calcd. for C15H20O3Na 271.1310, found 271.1307, ∆ = 1.1 ppm.7-(Benzyloxy)-methyl-2-(Z)-hepten-1-ol 17BnO17A solution of enoate 16 (2.86 g, 11.5 mmol) in THF (150 mL) was cooled to -78 ºC and DIBAL-H(28.8 mL of 1M solution in toluene, 28.8 mmol) was added dropwise over a period of 10 min. The resulting solution was stirred at -78 ºC for an additional 45 min and then warmed to 0 ºC. After 1.5 h, the reaction was quenched with aqueous saturated Rochelle’s salt (100 mL). The mixture was warmed to rt and stirred for an additional 1.5 h before the solution was extracted with EtOAc (3 x 60 mL). The combined organic phases were washed with H2O, saturated aqueous NaHCO3 and brine (60 mL each), dried over MgSO4, filtered and concentrated. Silica gel column chromatography (40% MTBE in hexanes) yielded 7-(benzyloxy)-methyl-2-(Z)-hepten-1-ol 17 (2.39 g, 94%) as a pale yellow oil.R f = 0.33 (50 % EtOAc in hexanes, UV/vanillin); IR (thin film, cm-1) 3369, 2935, 2859, 1454, 1102, 1027, 735, 697; 1H NMR (400 MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.65-5.50 (m, 2H), 4.50 (s, 2H), 4.18 (d, 2H, J = 6.6 Hz), 3.47 (t, 2H, J = 6.4 Hz), 2.10 (q, 2H, J = 7.3 Hz), 1.66-1.59 (m, 2H), 1.50-1.43 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 138.6, 132.8, 128.7, 128.3, 127.6, 127.5, 72.9, 70.2, 58.5, 29.2, 27.2, 26.2; HRMS (ESI) calcd. for C14H20O2Na 243.1361, found 243.1360, ∆ = 0.4 ppm.Epoxide 1818To a stirred 0 ºC solution of crushed 4Å molecular sieves (0.6 g) in CH2Cl2 (20 mL) were added D-(-)-diethyltartrate (0.23 mL, 0.28 g, 1.36 mmol) and Ti(O i Pr)4 (0.27 mL, 0.26 g, 0.91 mmol). This mixture was cooled to -20 ºC and tert-butyl hydroperoxide (1.8 mL of a ~5.0 M solution in isooctane, ~9.08 mmol) was added. The resulting mixture was stirred for 20 min before a solution of allylic alcohol 17 (1.0 g, 4.54 mmol) in CH2Cl2 (5 mL) was added dropwise via cannula. The allylic alcohol was first azeotropically dried with toluene and then dissolved in CH2Cl2 and dried over 4Å molecular sieves. The resulting mixture was stirred at -20 ºC for an additional 70 h and then warmed to 0 ºC. H2O (4 mL) wasadded and the stirring was continued at 0 ºC for an additional 1 h. The mixture was allowed to warm to rt.A solution of 30% NaOH in saturated aqueous NaCl (1 mL) was added and the resulting mixture was stirred for 45 min. The solution was diluted with H2O (4 mL) and filtered to get a better separation of the two phases. The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 x 20 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (40% ethyl acetate in hexanes) yielded epoxide 18 (0.67 g, 62%, 83% ee) as a colorless oil.R f= 0.27 (70 % EtOAc in hexanes UV/vanillin); [α]D = +2.5 (c 0.59, CH2Cl2); IR (thin film, cm-1) 3436, 2932, 2861, 1727, 1454, 1101, 1044, 737, 698; 1H NMR (400 MHz, CDCl3) δ 7.37-7.27 (m, 5H), 4.50 (s, 2H), 3.82 (ddd, 1H, J = 4.5 Hz, 7.4 Hz, 12.0 Hz), 3.68 (ddd, 1H, J = 5.1 Hz, 6.8 Hz, 12.0 Hz), 3.49 (t, 2H, J = 6.2 Hz), 3.14 (dt, 1H, J = 4.3 Hz, 6.8 Hz), 3.03 (m, 1H), 1.71-1.51 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 138.5, 128.4, 127.7, 127.6, 73.0, 70.0, 60.8, 57.2, 56.6, 29.4, 27.7, 23.5. These data match those reported in literature.3 HRMS (ESI) calcd. for C15H20O3Na 259.1310, found 259.1336, ∆ = 10 ppm. The enantiomeric purity was determined by HPLC (Daicel Chiralcel AD column, 15 % i-PrOH/hexanes, flow rate 0.5 mL/min): τmajor = 14.94 min; τminor = 16.85 min.7-(Benzyloxy)-2-methyl-heptane-1,3-diol 1919A mixture of CuCN (1.25 g, 14.0 mmol) in Et2O (35 mL) was cooled to -78 ºC and methyllithium(17.0 mL of a 1.48 M solution in Et2O, 25.1 mmol) was added dropwise. The resulting mixture was warmed to -20 ºC and stirred vigorously for 1 h. The color of the solution turned pale green (some CuCN remained undissolved). A solution of epoxide 18 (0.55 g, 2.33 mmol) in Et2O (15 mL) was added via cannula and the stirring was continued at -15 ºC for 2.5 h. Et2O (15 mL) and saturated aqueous NH4Cl (20 mL) were added. The resulting mixture was warmed to rt and filtered. The separated organic phase was washed with saturated aqueous NH4Cl, H2O and brine (2 x 25 mL each). The combined aqueous phases were back-extracted with Et2O (25 mL) and the combined organic phases were dried over Na2SO4, filtered and concentrated to yield the crude 19. To this crude material were added THF (15 mL), H2O (15 mL) and NaIO4 (0.16 g, 0.77 mmol) at rt. The resulting solution was stirred for 1.5 h and then diluted with Et2O (20 mL). The separated organic phase was washed with saturated aqueous NaHCO3 (25 mL) and brine (25 mL). The combined aqueous phases were back-extracted with Et2O (25 mL) and the combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (50% MTBE in hexanes), followed by recrystallization of the product from EtOAc/hexanes (1:10) yielded diol 19 (0.38 g, 65%) as white crystalline solid.R f= 0.23 (70 % EtOAc in hexanes, UV/vanillin); mp 50-52 ºC; [α]D = -5.7 (c 0.42, CH2Cl2) (95 % ee); IR (thin film, cm-1) 3370, 2937, 2864, 1455, 1101, 1028, 736, 698; 1H NMR (400 MHz, CDCl3) δ 7.37-7.26 (m, 5H), 4.50 (s, 2H), 3.82 (m, 1H), 3.69 (m, 2H), 3.49 (dt, 2H, J = 0.9 Hz, 12.7 Hz), 2.22 (s, 2H), 1.82-1.36 (m, 7H), 0.9 (d, 3H, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ 138.5, 128.4, 127.7, 127.5, 74.5, 72.9, 70.3, 67.2, 39.1, 33.8, 29.6, 22.9, 10.1; These data match those reported in literature3; HRMS (ESI) calcd. for C15H24O3Na 275.1623, found 275.1628, ∆ = 1.8 ppm; Anal. calcd. for C15H24O3: C, 71.39; H, 9.59; found: C, 71.62; H, 9.64.7-(Benzyloxy)-2-methyl-1-(tert-butyl-diphenylsilyl)-heptan-3-olTo a stirred 0 ºC solution of diol 19 (0.38 g, 1.51 mmol) in CH2Cl2 (6 mL) were added triethylamine (0.48 mL, 1.35 g, 3.46 mmol) and TBDPSCl (0.47 mL, 0.5 g, 1.81 mmol). After additions the mixture was allowed to warm to rt and stirring was continued for an additional 14 h. The mixture was diluted with Et2O (5 mL) and washed with H2O (5 mL). The aqueous phase was extracted with Et2O (5 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated. Silica gel column chromatography (30% EtOAc in hexanes) yielded 7-(benzyloxy)-2-methyl-1-(tert-butyl-diphenylsilyl)-heptan-3-ol (0.73 g, 98%) as a pale yellow viscous oil.R f= 0.69 (70 % EtOAc in hexanes, UV/vanillin); [α]D = -3.4 (c 0.44, CH2Cl2); IR (thin film, cm-1) 3460, 2932, 2858, 1472, 1428, 1112, 740, 702; 1H NMR (400 MHz, CDCl3) δ 7.68-7.65 (m, 4H), 7.46-7.27 (m, 11H), 4.51 (s, 2H), 3.86 (m, 1H), 3.75 (dd, 1H, J = 4.2 Hz, 10.1 Hz), 3.66 (dd, 1H, J = 6.0 Hz, 10.1 Hz), 3.49 (dt, 2H, J = 1.0 Hz, 6.5 Hz), 2.77 (d, 1H, J = 2.9 Hz), 1.79-1.36 (m, 7H), 1.06 (s, 9H), 0.90 (d, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 138.7, 135.7, 135.6, 133.1, 133.0, 129.83, 129.79, 128.3, 127.8, 127.6, 127.5, 74.1, 72.9, 70.4, 68.7, 39.1, 34.0, 29.8, 26.9, 22.9, 19.2, 10.2; HRMS (ESI) calcd. for C31H42O3NaSi 513.2801, found 513.2823, ∆ = 4.3 ppm.-butyl-diphenylsilyl)-heptane-1,5-diol 206-Methyl-7-(tertTo a stirred solution of 7-(benzyloxy)-2-methyl-1-(tert-butyl-diphenylsilyl)-heptan-3-ol (0.66 g, 1.34 mmol) in EtOAc (20 mL) was added Pd(OH)2 on charcoal (0.10 g of 20% Pd catalyst, 0.15 mmol) under argon flow. The reaction flask was repeatedly evacuated and flushed with H2. The suspension was vigorously stirred under H2 atmosphere for 13 h and then filtered through Celite. The filter pad was washed with EtOAc (3 x 20 mL) and the combined filtrates were concentrated. Silica gel columnchromatography (40% EtOAc in hexanes) yielded 6-methyl-7-(tert-butyl-diphenylsilyl)-heptane-1,5-diol 20 (0.46 g, 89%) as a colorless viscous oil.R f= 0.33 (70 % EtOAc in hexanes, UV/vanillin); [α]D = -3.4 (c 0.61, CH2Cl2); IR (thin film, cm-1) 3339, 2931, 2585, 1428, 1112, 740, 701; 1H NMR (400 MHz, CDCl3) δ 7.68-7.65 (m, 4H), 7.46-7.38 (m, 6H), 3.87 (m, 1H), 3.76 (dd, 1H, J = 4.2 Hz, 10.1 Hz), 3.69-3.65 (m, 3H), 2.02 (br s, 2H), 1.80-1.71 (m, 1H), 1.66-1.35 (m, 6H), 1.06 (s, 9H), 0.92 (d, 3H, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.6, 133.1, 132.9, 129.9, 129.8, 127.8, 74.2, 68.7, 62.9, 39.2, 33.8, 32.7, 26.9, 22.5, 19.2, 10.3; HRMS (ESI) calcd. for C24H36O3NaSi 423.2331, found 423.2345, ∆ = 3.3 ppm. The enantiomeric purity was determined by HPLC (Daicel Chiralcel OD column, 1:9 i-PrOH/hexanes, flow rate 0.5 ml/min): τmajor = 11.15 min; τminor = 14.82 min, 95% ee.Lactone 21To a stirred solution of diol 20 (0.70 g, 1.75 mmol) in CH2Cl2 (20 mL) were added crushed 4 Å molecular sieves (0.75 g) and pyridinium chlorochromate (PCC, 0.66 g, 3.06 mmol) at rt. After 3 h, second portions of molecular sieves (0.25 g) and PCC (0.66 g, 3.06 mmol) were added. Stirring was continued for an additional 13 h and a third portion molecular sieves (0.2 g) and PCC (0.57 g, 2.63 mmol) were added. After 3 h, a third portion of PCC (0.18 g, 1.88 mmol) was added. This mixture was stirred for an additional 3 h before the mixture was filtered through silica gel with EtOAc and then concentrated. Silica gel column chromatography (10% EtOAc in hexanes) yielded lactone 21 (0.40 g, 58%) as a colorless oil.R f= 0.56 (70 % EtOAc in hexanes, UV/vanillin); [α]D = +26.3 (c 1.15, CH2Cl2); IR (thin film, cm-1) 2958, 2931, 2883, 2857, 1736, 1428, 1239, 1112, 702; 1H NMR (400 MHz, CDCl3) δ 7.67-7.63 (m, 4H), 7.45-7.37 (m, 6H), 4.49 (ddd, 1H, J = 3.0 Hz, 4.1 Hz, 11.5 Hz), 3.73 (dd, 1H, J = 7.0 Hz, 10.2 Hz), 3.61 (dd, 1H, J = 5.3 Hz, 10.2 Hz), 2.58 (m, 1H), 2.41 (dt, 1H, J = 7.2 Hz, 8.8 Hz), 1.96-1.77 (m, 4H), 1.70-1.59 (m, 1H), 0.98 (s, 9H), 0.97 (d, 3H, J = 6.9 Hz); 13C NMR (100 MHz, CDCl3) δ 172.0, 135.6, 135.5, 133.7, 133.5, 129.7, 127.7, 80.4, 65.1, 40.4, 29.5, 26.9, 25.5, 19.3, 18.7, 11.1; HRMS (ESI) calcd. for C24H32O3NaSi 419.2018, found 419.2011, ∆ = 1.6 ppm.Ketoalcohol 22OH2222bTo a stirred mixture of Mg powder (36 mg, 1.46 mmol) in THF (3 mL) was added 4-bromo-1-butene (0.15 mL, 1.19 g, 1.46 mmol). Heat was evolved and the formation of the Grignard reagent was evident from the darkening of the reaction mixture. After 50 min, 1.55 mL of the solution of the Grignard reagent prepared above was added dropwise to a -78 ºC solution of lactone 21 (0.2 g, 0.504 mmol) in THF (3 mL). The resulting solution was stirred at -78 ºC for an additional 40 min and then saturated aqueous NH4Cl (1.5 mL) and H2O (1.5 mL) were added. The mixture was warmed to rt and then extracted with MTBE (3 x 10 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. The crude product was purified twice with flash chromatography (20 % EtOAc in hexanes, 15% EtOAc in hexanes) to yield ketoalcohol 22 (60.7 mg, 27%) as a pale yellow oil and lactone 21 (132.8 mg, 66%). This process was repeated twice to obtain 22 in 60% overall yield.R f= 0.44 (40 % EtOAc in hexanes, UV/vanillin); [α]D = +0.5 (c 0.42, CH2Cl2); IR (thin film, cm-1) 3469, 2930, 2857, 1713, 1428, 1275, 1261, 1112, 912, 824, 750, 702; 1H NMR (400 MHz, CDCl3) For 22: δ 7.69-7.66 (m, 4H), 7.48-7.39 (m, 6H), 5.82 (ddt, 1H, J = 17.1 Hz, 10.3 Hz, 6.6 Hz), 5.01 (dq, 1H, J = 17.1 Hz, 1.8 Hz), 4.94 (dtd, 1H, J = 2.0 Hz, 1.3 Hz, 10.3 Hz), 3.72-3.68 (m, 1H), 3.67 (dd, 1H, J = 6.7 Hz, 10.0 Hz), 3.57 (dd, 1H, J = 5.7 Hz, 9.8 Hz), 2.62 (d, 1H, J = 5.5 Hz), 2.49 (t, 2H, J = 7.3 Hz), 2.41 (t, 2H, J = 7.3 Hz), 2.28-2.22 (m, 2H), 1.78-1.27 (m, 5H), 1.03 (s, 9H), 0.84 (d, 3H, J = 7.0 Hz); For 22b: δ 5.86 (m), 4.99 (dq, J = 17.2 Hz, 5.5 Hz), 4.90 (m), 3.56 (dd), 0.95 (d, 3H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) For 22: δ 211.0, 136.44, 136.40, 134.7, 134.6, 130.8, 128.75, 115.3, 71.9, 67.9, 43.0, 42.2, 41.4, 34.8, 28.8, 27.2, 21.2, 19.8, 10.9; For 22b: δ 140.3, 138.7, 130.7, 128.70, 114.5, 96.9, 71.0, 67.0, 43.1, 41.7, 34.0, 28.8, 28.4, 20.0, 12.7; HRMS (ESI) calcd. for C28H40O3NaSi 475.2644, found 475.2667, ∆ = 4.8 ppm.Ketal 2323To a stirred solution of ketoalcohol 22 (50 mg, 0.11 mmol) in CH2Cl2 (4 mL) at rt was added p-methoxybenzyl alcohol (0.1 mL, 0.11 g, 0.77 mmol) and pyridinium p-toluenesulfonate (5.5 mg, 0.022 mmol). The resulting solution was stirred for 3 h and then saturated aqueous NaHCO3 (2 mL) and H2O (4 mL) were added. The separated aqueous phase was further extracted with EtOAc (4 x 10 mL) and the combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel columnchromatography (3% MTBE in hexanes, silica gel containing ca. 0.1% Ca (Fluka)) yielded ketal 23 (53 mg, 83%) as a pale yellow oil.R f= 0.56 (60 % EtOAc in hexanes, UV/vanillin); [α]D = +7.4 (c 1.04, CH2Cl2); IR (thin film, cm-1) 3070, 2958, 2931, 2857, 1726, 1472, 1428, 1239, 1112, 1027, 931, 824, 741, 702; 1H NMR (400 MHz, CDCl3) δ 7.65-7.63 (m, 4H), 7.46-7.36 (m, 6H), 7.23-7.20 (m, 2H), 6.82-6.78 (m, 2H), 5.86 (ddt, 1H, J = 17.1 Hz, 10.3 Hz, 6.6 Hz), 5.02 (dq, 1H, J = 17.1 Hz, 1.7 Hz), 4.93 (dtd, 1H, J = 1.2 Hz, 1.6 Hz, 10.3 Hz), 4.36 (d, 1H, J = 11.0 Hz), 4.27 (d, 1H, J = 11.2 Hz), 3.78- 3.76 (m, 1H), 3.74 (s, 3H), 3.72 (dd, 1H, J = 5.1 Hz), 3.61 (dd, 1H, J = 6.0 Hz, 10.0 Hz), 2.11-2.02 (m, 2H), 1.85-1.16 (m, 9H), 1.01 (d, 3H), 1.00 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.9, 139.9 136.4, 134.84, 134.82, 132.2, 130.71, 130.69, 130.0, 128.7, 114.7, 114.5, 100.4, 71.8, 67.0, 61.9, 55.8, 41.8, 37.3, 33.4, 28.8, 28.7, 27.3, 19.9, 19.8, 12.8; HRMS (ESI) calcd. for C36H48O4NaSi 595.3220, found 595.3209, ∆ = 1.8 ppm.Dihydroxyketal 2424b24a(DHQ)2PYR (5.6 mg, 0.006 mmol), K3Fe(CN)6 (89 mg, 0.27 mmol), K2CO3 (37 mg, 0.27 mmol), CH3SO2NH2 (8.6 mg, 0.09 mmol) and K2OsO4·2H2O (0.3 mg, 0.0009 mmol) were dissolved in 1:1 tert-butyl alcohol/water (1.5 mL each) at rt. The resulting mixture was vigorously stirred for 20 min and then cooled to 0 ºC. A solution of ketal 23 (52 mg, 0.09 mmol) in tert-butyl alcohol (0.5 mL) was added via cannula. Stirring was continued at 0 ºC for an additional 17.5 h before Na2SO3 (0.14 g) was added. The resulting mixture was vigorously stirred and allowed to warm to rt. The mixture was diluted with EtOAc (10 mL). Separated aqueous phase was extracted with EtOAc (3 x 5 mL) and the combined organic phases were washed with H2O (10 mL), dried over Na2SO4, filtered and concentrated. The crude product was immediately used in the next reaction without further purification.R f= 0.16 (60 % EtOAc in hexanes, UV/vanillin); 1H NMR (400 MHz, CDCl3) δ 7.65-7.63 (m, 4H), 7.47- 7.37 (m, 6H), 7.23-7.20 (m, 2H), 6.81-6.79 (m, 2H), 4.36-4.26 (m, 2H), 3.77-3.76 (m, 1H), 3.74 (s, 3H), 3.72 (dd, 1H, J = 5.0 Hz), 3.61 (dd, 1H, J = 5.8 Hz, 9.9 Hz), 3.49 (m, 1H), 3.41 (m, 1H), 3.30 (m, 1H), 2.82 (m, 1H), 2.69 (m, 1H), 1.81-1.23 (m, 11H), 1.01 (d, 3H), 1.00 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 159.8, 136.4, 134.82, 134.80, 132.3, 130.69, 130.68, 130.0, 128.7, 114.5, 100.5, 72.8, 71.8, 67.1, 67.0, 61.8, 55.8, 41.8, 34.1, 33.4, 28.8, 28.2, 27.3, 19.9, 19.8, 12.8; For 24b, the following signals were also observed in 13C NMR: δ 72.8, 67.1, 61.9, 34.2, 28.9, 28.1.Pivalate 2525a25bTo a stirred 0 ºC solution of dihydroxyketal 24 (0.05 g, 0.082 mmol) in pyridine (0.5 mL) was added pivaloyl chloride (12 µl, 12 mg, 0.1 mmol). The resulting solution was stirred at 0 ºC for an additional 2 h 15 min. Aqueous saturated NaHCO3 (1 mL) and H2O (5 mL) were added and the phases were separated. The aqueous phase was extracted with EtOAc (3 x 5 mL) and the combined organic phases were washed with H2O (5 mL), dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (15% EtOAc in hexanes) yielded pivalate 25 (40.1 mg, 62% over 2 steps) as a pale yellow viscous oil.R f= 0.47 (40 % EtOAc in hexanes, UV/vanillin); [α]D = +12.4 (c 0.27, CH2Cl2); IR (thin film, cm-1) 3469, 2930, 2957, 1713, 1428, 1275, 1261, 1112, 912, 824, 750, 701; 1H NMR (400 MHz, CDCl3) For 25a: δ7.65-7.62 (m, 4H), 7.46-7.36 (m, 6H), 7.22-7.20 (m, 2H), 6.80-6.78 (m, 2H), 4.35 (dd, 1H, J = 3.0 Hz, 11.1 Hz), 4.28 (dd, 1H, J = 4.6 Hz, 11.1 Hz), 3.96-3.91 (m, 2H), 3.77-3.76 (m, 1H), 3.74 (s, 3H), 3.72 (dd, 1H, J = 4.9 Hz), 3.61 (dd, 1H, J = 5.9 Hz, 10.0 Hz), 2.97 (d, 1H, J = 5.5 Hz), 1.80-1.21 (m, 11H), 1.17 (s, 9H), 1.01 (dd, 3H), 1.00 (s, 9H); For 25b, the following additional resonances could be observed: δ 2.97 (d, J = 5.3 Hz), 1.17 (s); 13C NMR (100 MHz, CDCl3) For 25a: δ 178.93, 159.8, 136.4, 134.83, 134.81, 132.3, 130.70, 130.69, 130.0, 129.9, 128.7, 114.5, 100.4, 71.8, 70.1, 68.9, 67.0, 61.85, 55.8, 41.9, 39.4, 34.0, 33.4, 28.8, 28.4, 27.4, 27.2, 19.9, 19.8, 12.8; For 25b, the following additional resonances could be observed: δ 178.91, 100.5, 70.0, 68.8, 61.89, 28.5; HRMS (ESI) calcd. for C41H58O7NaSi 713.3850, found 713.3872, ∆ = 3.0 ppm.Spiroketals 12 and 261226To a solution of pivalate 25 (3.5 mg, 0.005 mmol) in CDCl3 (1 mL) was added AcOH (1 drop, ca 10 µL, 3300 mol%) at rt. After 21 h, the reaction was quenched by the addition of saturated aqueous NaHCO3 (0.5 mL) and H2O (0.5 mL). The separated aqueous phase was extracted with EtOAc (3 x 3 mL) and the combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3% EtOAc in hexanes) yielded fraction A: spiroketal 12 (1.5 mg, 54%), fraction B: a mixture of spiroketals 12 and 26 (0.5 mg, 17%), fraction C: spiroketal 26 (0.8 mg, 29%).12: R f= 0.33 (10 % EtOAc in hexanes, UV/vanillin); [α]D = +2.6 (c 0.53, CH2Cl2); IR (thin film, cm-1) 2930, 2857, 1732, 1460, 1367, 1282, 1153, 1113, 1075, 1023, 824, 742, 702; 1H NMR (400 MHz, CDCl3) δ 7.67-7.64 (m, 4H), 7.48-7.38 (m, 6H), 4.19-4.12 (m, 1H), 4.03 (dd, 1H, J = 3.9 Hz, 11.4 Hz), 3.97 (dd, 1H, J = 5.0 Hz, 11.4 Hz), 3.83 (ddd, 1H, J = 2.1 Hz, 5.3 Hz, 11.7 Hz), 3.66 (dd, 1H, J = 6.0 Hz, 9.9 Hz), 3.52 (dd, 1H, J = 6.0 Hz, 9.9 Hz), 2.05-1.98 (m, 1H), 1.79-1.57 (m, 7H), 1.49-1.44 (m, 1H), 1.36-1.22 (m, 2H), 1.15 (s, 9H), 1.04 (s, 9H), 0,90 (d, 3H, J = 7.0 Hz); 13C NMR (100 MHz, CDCl3) δ 178.8, 136.41, 136.39, 134.89, 134.84, 130.7, 128.7, 107.2, 76.3, 71.4, 67.0, 66.7, 41.4, 39.4, 38.1, 33.5, 28.7, 27.4, 27.3, 26.6, 21.4, 19.8, 12.4; HRMS (ESI) calcd. for C33H48O5NaSi 575.3169, found 575.3176, ∆ = 1.2 ppm.26: R f= 0.30 (10 % EtOAc in hexanes, UV/vanillin); [α]D = +1.4 (c 0.14, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.67-7.64 (m, 4H), 7.47-7.38 (m, 6H), 4.21- 4.13 (m, 1H), 3.99 (dd, 1H, J = 7.3 Hz, 11.0 Hz), 3.94 (dd, 1H, J = 5.3 Hz, 11.0 Hz), 3.73 (ddd, 1H, J = 2.1 Hz, 5.7 Hz), 3.70 (dd, 1H, J = 4.6 Hz, 9.9 Hz), 3.54 (dd, 1H, J = 6.9 Hz, 9.9 Hz), 2.11-2.10 (m, 1H), 1.86-1.47 (m, 10H), 1.15 (s, 9H), 1.03 (s, 9H), 0,97 (d, 3H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 178.7, 136.4, 134.9, 134.8, 130.69, 130.67, 128.7, 107.3, 78.3, 72.2, 69.0, 67.1, 41.9, 39.0 (2 C), 34.1, 28.6, 27.8, 27.4, 27.3, 21.2, 19.8, 13.4. HRMS (ESI) calcd. for C33H48O5NaSi 575.3169, found 575.3157, ∆ = 2.1 ppm.Spiroketal 111127To a solution of pivalate 25 (10.0 mg, 0.014 mmol) in CH2Cl2 (1 mL) was added chloroacetic acid (0.27 mg, 0.0029 mmol) in CH2Cl2 (0.8 ml) at rt. After 30 min, another portion of chloroacetic acid (0.27 mg, 0.0029 mmol) in CH2Cl2 (0.8 ml) was added. The resulting solution was stirred at rt for an additional 4h. The reaction was quenched by addition of saturated aqueous NaHCO3 (0.5 mL) and H2O (0.5 mL). The separated aqueous phase was extracted with EtOAc (3 x 5 mL) and the combined organic phases were dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (3% EtOAc in hexanes) yielded fraction A: spiroketal 12 (1.9 mg, 24%), fraction B: a mixture of spiroketals 12 and 26 (1.6 mg, 21%), fraction C: spiroketal 26 (0.5 mg, 6%), fraction D: spiroketal 11 (containing ca 5% of 27) (3.8 mg, 49%).Fraction D: R f= 0.20 (10 % EtOAc in hexanes, UV/vanillin); [α]D = +5.4 (c 0.28, CH2Cl2); IR (thin film, cm-1) 2929, 2857, 1732, 1463, 1277, 1263, 1156, 1113, 1009, 896, 824, 748, 703; 1H NMR (400 MHz, CDCl3) δ 7.68-7.65 (m, 4H), 7.48-7.39 (m, 6H), 4.20-4.14 (dt, J = 4.6 Hz, 7.3 Hz), 4.02 (dd, 1H, J = 4.6 Hz, 11.1 Hz), 3.92 (dd, 1H, J = 7.3 Hz, 11.1 Hz), 3.67 (dd, 1H, J = 6.8 Hz, 9.9 Hz), 3.63-3.58 (ddd, 1H, J = 2.3 Hz, 4.5 Hz, 11.5 Hz), 3.52 (dd, 1H, J = 6.0 Hz, 9.9 Hz), 2.42-2.37 (m, 1H), 1.79-1.24 (m, 10H), 1.17 (s, 9H), 1.04 (s, 9H), 0.89 (d, 3H, J = 6.9 Hz); 13C NMR (100 MHz, CDCl3) δ 178.7, 136.4, 134.8,130.7, 128.7, 109.4, 78.1, 75.0, 69.2, 67.0, 41.4, 39.3, 34.8, 32.2, 28.9, 27.6, 27.4, 27.2, 22.9, 19.8, 12.3; HRMS (ESI) calcd. for C 33H 48O 5NaSi 575.3169, found 575.3159, ∆ = 1.7 ppm.Additional evidence for the assigned nonanomeric structure of 11 (Figure 3). 1H NMR coupling constant data clearly indicates a C4 chair conformation for the A ring.11J H3-H4a 11.5 Hz J H3-H4e 2.3 HzSpiroketalization with PPTS (Table 1, entry 1) afforded 11 accompanied by ca. 10% of the inseparable C 10 epimer 27. For spiroketal 27, the following additional resonances could be observed: δ 4.29-4.22 (m), 4.05 (dd, J = 3.8 Hz), 3.95 (dd, J = 5.1 Hz), 3.69-3.65 (m), 1.18 (s), 0.89 (d, J = 6.8 Hz).References1. Gao, Y.; Hanson, R. M.; Klunder, J. M.; Ko, S. Y.; Masamune, H.; Sharpless, K. B. J. Am. Chem. Soc. 1987, 109, 5765-5780.2. Börjesson, L.; Csöregh, I.; Welch, C. J. J. Org. Chem. 1995, 60, 2989-2999.3. Nicolaou, K. C.; Veale, C. A.; Webber, S. E.; Katerinopoulos, H. J. Am. Chem. Soc. 1985, 107, 7515- 7518.1.04516.25596.23716.22716.21846.20836.1896(ppm)6.202.17183.50423.48863.4721(ppm)3.502.12092.71132.70722.69252.68842.67382.66972.65552.6509(ppm)2.702.19182.79161.70401.70171.68521.67061.64821.63351.60201.57141.56221.54441.52521.50781.5051(ppm)1.601.705.48141.04511.01832.09283.07342.17182.12092.19182.7916I n t e g r a l7.36617.36337.34967.34147.33187.31997.30537.29437.28427.27337.26697.26007.25136.25596.23716.22716.21846.20836.18965.79635.79225.78815.76805.76345.75934.50103.69893.50423.48863.47212.71132.70722.69252.68842.67382.66972.65552.65091.70401.70171.68521.67061.64821.63351.60201.57141.56221.54441.52521.50781.5051(ppm)jea20791.01835.79635.79225.78815.76805.76345.7593(ppm) 5.76166.7723150.4313138.6004128.3098127.5714127.4523119.416877.317677.000076.682472.855270.020550.940129.318928.659825.6028(ppm)jea2079。


苯基硼酸化学式苯基硼酸,也称为硼酸苯,是一种有机化合物,化学式为C6H5B(OH)3。
它是一种无色晶体,然而在水中它具有淡黄色至深黄色的溶液。
苯基硼酸是一种有机酸,溶于水和乙醇。
其结构包含一个苯环,上有一个羟基,每个羟基由一个硼原子键合。
苯基硼酸的物理性质和溶解特性取决于它的结构。
由于苯环具有较高的分子间相互作用力,苯基硼酸具有很高的沸点,且极易溶于水和有机溶剂,因此具有较高的溶解性。
苯基硼酸的合成方法有很多种,其中最常用的是以硼酸和苯为原料,经过水解反应生成。
在室温下,硼酸和苯发生反应,形成六元环结构的苯基硼酸。
硼酸和苯的反应如下:2C6H5OH + B(OH)4 = C6H5B(OH)3 + 2H2O其中,B(OH)4表示硼酸的负离子,即tan [B(OH)4],而C6H5B(OH)3表示苯基硼酸。
此外,苯基硼酸也可以通过电极反应来合成,其合成反应如下: 2H2 + 2e- 2H2O2B + 6e- 2B(OH)4-C6H5OH + 2B(OH)4- C6H5B(OH)3 + 2H2O可见,苯基硼酸的合成反应是由硼酸和苯酚构成的水解反应,或者是以水和电极反应构成的电解反应。
苯基硼酸的化学性质非常有趣。
它是一种有机酸,具有较强的酸性,溶解性也很强,而且易于水解反应,因此它可以用来制备其他有机酸,也可以用来形成混合离子溶液。
另外,苯基硼酸也可以用作自由基反应的催化剂,以及用于合成有机化合物的原料。
总之,苯基硼酸是一种重要而有趣的有机化合物,能够发挥多种作用,如制备其他有机酸、形成混合离子溶液、作为自由基反应催化剂和用于合成有机化合物原料等。
其化学式为C6H5B(OH)3。


2-ph和iph高碳醇2-phenylethanol和isopropylphenyl高碳醇是常用的赋香醇,常用于化妆品、食品、香料和药品中。
2-phenylethanol是一种独特的异构体,它被广泛应用于香水、乳液、护肤霜、洗发水、肥皂、香精和药品中。
它是一种具有微弱玫瑰般香味的无色液体。
它是可溶于水的,但不易挥发。
2-phenylethanol由苯基乙醇和其它化合物混合而成。
它一般具有强烈的花香、玫瑰香、草香或者甜味。
它是一种天然多酚类化合物,能够起到抗氧化和抗炎作用,它也是一种健康的食品添加剂。
它常被用于卫生护理领域,例如香水、护肤霜、肥皂和洗发水中。
2-phenylethanol还在缓解抑郁症、焦虑症和其他情绪障碍等领域具有潜在的医疗用途。
Isopropylphenyl高碳醇是一种受欢迎的合成香,它被广泛应用于化妆品、香水、洗发水、肥皂和其他非食品领域。
它是一种具有清新、田园和海洋香味的油状液体。
它可以通过化学反应获得,例如苯乙烯和异丙醇在催化剂条件下的反应。
这种化合物通常以1-10%的浓度混合在化妆品、香水和肥皂等产品中。
它可以作为植物调香剂,用于替代天然芳香剂。
它也具有一定的杀菌和抗炎作用,因此被广泛应用于护肤产品和医疗产品领域。
总之,2-phenylethanol和isopropylphenyl高碳醇是两种重要的赋香醇,它们被广泛应用于化妆品、香水、食品和药品等领域。
它们具有独特而令人愉悦的香味,同时也具有其他功效,如抗氧化和抗炎作用等,因此被广泛应用于各种领域,成为人们日常生活中不可或缺的一部分。

苯基正丁基醚
苯基正丁基醚是一种常见的有机合成原料,具有水溶性、高稳定性和有效位价
等特点,可广泛应用于高分子材料的合成、染料、农药、药物中间体、复配助剂、燃料添加剂、润滑油添加剂等多种产品的制备。
苯基正丁基醚是一种有机溶剂,用作水溶性涂料溶剂或溶剂溶解其它有机物质,可用于精细化学品的制造。
它可以清除细胞膜的有机污染或残留物,促进微小粒子的吸附等。
由于具有极低的析出热和极好的蒸汽压,经常作为延长油里程的添加剂应用于汽油机燃油中。
正丁基醚在新材料。
高分子材料中也具有极广泛的应用,如制备合成橡胶、醋
酸吡咯类材料、酯类聚合物、高分子醚、聚维酮和改性粘合剂等。
此外,它在生物工程中也被用作原料和溶剂,如催化化学酶的合成,还可用作基础溶剂和表面活性剂,并可用于改进涂料的稳定性、柔性和耐候性。
正丁基醚的特性,使其广泛用于工业和农业生产中,适用于制造大量的有机化
学品,如染料、香料、蔬菜、水果、调味品、保健品等,也适用于农药、植物生长调节剂等。
此外,它还可用作一种工业催化剂,用于合成氨酸、醛或酯,酸类及其他类型的有机化合物。
对于蔬菜中的病虫害还有一定的防治效果,但毒性相对较高,使用前应当适当加以考虑和考量。
苯基正丁基醚具有高稳定性,固体熔点低,易溶于水和常见有机溶剂,安全和
可靠等优点,已被广泛应用于各行各业。
此外,由于具备更高的耐热性和耐电导率特性,成为乙烯叙氧化物绝缘液中所必需的中间体。
有利于更多有机材料的发展。

CHEMLIST, CHEMCATS: Copyright ?2009 American Chemical Society. All Rights Reserved.CASREACT: Copyright ?2009 American Chemical Society. All Rights Reserved. CASREACT contains reactions from CAS and from: ZIC/VINITI database (1974-1999) provided by InfoChem; INPI data prior to 1986;Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich; organic reactions, portions copyright 1996-2006 John Wiley & Sons, Ltd., John Wiley and Sons, Inc., Organic Reactions Inc., and Organic Syntheses Inc. Reproduced under license. All Rights Reserved.REGISTRY: Copyright ?2009 American Chemical Society. All Rights Reserved. (Some records contain information from GenBank(R). See also: Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Rapp B.A., Wheeler D.L. Genbank. Nucl. Acids Res. 28(1):15-18 (2000). Property values tagged with IC are from the ZIC/VINITI data file provided by InfoChem.) CAS Registry is a service mark of the American Chemical Society.MEDLINE: Produced by the U.S. National Library of MedicineCAPLUS: Copyright ?2009 American Chemical Society. All Rights Reserved. (The UK patent material in thisproduct/service is UK Crown copyright and is made available with permission. ?Crown Copyright. The French (FR) patent material in this product/service is made available from Institut National de la Propriete Industrielle (INPI).)Copyrights:6 Reactions.Explored Reactions containing Registry Number 71544-94-0 in any role in CASREACT Selected scores: ≥ 99 (most similar).Analyze by Similarity started.10533 SubstancesExplored for similar substances in REGISTRY.Get substances with structures similar to this structure OOOOOOPInput structure:Structure task started on Thu Feb 25, 2010 at 1:13 PM Task History(Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from theCASREACTZhurnal Obshchei Khimii, 49(7), 1474-91; 1979OMeMeOClCHCO +MeOPCl 2+PhOH(Step 2.1)2 C:P(OEt)31 R:FeCl 3OMe OMePhOOPhPCHOC ONOTE: Reactants: 3, Reagents: 1, Catalysts: 1, Steps: 2, Stages: 2Answer 3: (Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from the following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)CASREACTZhurnal Obshchei Khimii, 49(7), 1474-91; 1979EtOH +OMe OMe FFCHC P OO+PhOH (Step 3.1)3 C:P(OEt)32 R:PCl 51 C:P(OEt)3OMe OMePhOOPhP CHOC ONOTE: Reactants: 3, Reagents: 1, Catalysts: 1, Steps: 3, Stages: 3Answer 2: (Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from the following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)CASREACTZhurnal Obshchei Khimii, 49(7), 1474-91; 1979P(OEt)3+OMeMeOClCHCO +PhOH(Step 3.1)3 C:P(OEt)32 R:PCl 5OMe OMePhOOPhPCHOC ONOTE: Reactants: 3, Reagents: 1, Catalysts: 1, Steps: 3, Stages: 3Answer 1:PhOH +OMe OMe ClClCHC P OOC:P(OEt)3OMe OMePhOOPhPCHO C ONOTE: Reactants: 2, Catalysts: 1, Steps: 1, Stages: 1Answer 6: (Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from the following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)CASREACTZhurnal Obshchei Khimii, 49(7), 1474-91; 1979OMeMeOClCHCO +Cl 2POEt+PhOH(Step 2.1)2 C:P(OEt)31 R:FeCl 3OMe OMePhOOPhPCHOC ONOTE: Reactants: 3, Reagents: 1, Catalysts: 1, Steps: 2, Stages: 2Answer 5: (Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from the following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)CASREACTZhurnal Obshchei Khimii, 49(7), 1474-91; 1979EtOOEtOMe OMe CHPC OO+PhOH(Step 2.1)2 C:P(OEt)31 R:PCl 5OMe OMePhOOPhP CHO C ONOTE: Reactants: 2, Reagents: 1, Catalysts: 1, Steps: 2, Stages: 2Answer 4:following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)Zhurnal Obshchei Khimii, 49(7), 1474-91; 1979CASREACT(Copyright (C) 2011 ACS. In addition to reactions indexed by CAS, CASREACT contains reactions derived from the following sources: ZIC/VINITI database (1974-1999) provided by InfoChem, INPI data prior to 1986, and Biotransformations database compiled under the direction of Professor Dr. Klaus Kieslich.)。