7 阿片样镇痛药
- 格式:doc
- 大小:543.00 KB
- 文档页数:17
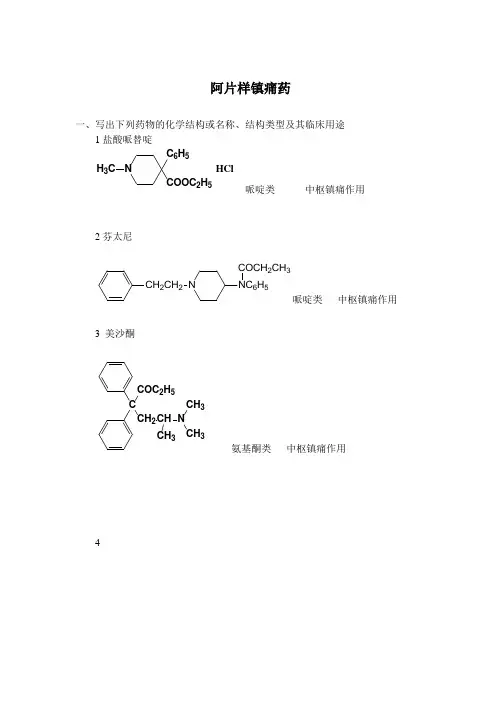
阿片样镇痛药一、写出下列药物的化学结构或名称、结构类型及其临床用途1盐酸哌替啶N COOC 2H 5C 6H 5H 3C HCl哌啶类 中枢镇痛作用2芬太尼CH 2CH 2NNC 6H 5COCH 2CH 3哌啶类 中枢镇痛作用3 美沙酮CCH 2COC 2H 5CH NCH 3CH 3CH 3氨基酮类 中枢镇痛作用43吗啡生物碱类中枢镇痛作用5CH3NCH3可待因生物碱类中枢镇咳作用62纳洛酮吗啡烃类吗啡中毒解救7镇痛新苯吗喃类非麻醉性中枢镇痛药物二、写出下列药物的结构通式1合成镇痛药N三、名词解释1. 阿片样镇痛药 是指以吗啡为代表的作用于中枢阿片受体,具有减轻锐痛和剧痛的作用的一类药物,有一定的成瘾性。
2.阿片受体 即阿片样物质受体,是指能与吗啡等阿片样物质结合产生强镇痛效果的生物大分子,存在于脑部、脊髓组织、外周神经系统等,分为μ、κ、δ三种。
四、简答题1、试写出合成镇痛药的结构类型并各举出一例药物名称。
答: 1、吗啡喃类:酒石酸那洛啡尔2、苯吗喃类:喷他佐辛3、哌啶类:盐酸哌替啶4、氨基酮类:盐酸美沙酮5、环己烷衍生物:盐酸曲马多6、氨基四氢奈类:地佐幸2、(1)完成反应式,写出最后产物的名称和主要用途CH 3NH 2O2CH 3NCH 2CH 2Cl CH 2CH 2ClC 6H 5CHCN H ,NaNH 224H 2ONH 3C C 6H 5COOC 2H 5HCl答:CH 3NH 2O3NCH 2CH 2OH CH 2CH 2OH 2CH 3NCH 2CH 2ClCH 2CH 2ClC 6H 5CHCN H ,NaNH 2N3C C 6H 5CN 24H 2ON 3C C 6H 5COOH25NH 3C C 6H 5COOC 2H 5HClN(H 3C C 6H 5COOC 2H 5HCl))))最后产物:盐酸哌替啶,为中枢镇痛药物,合成镇痛药物。
(2)写出该药物的化学名全称,并简要说明该药物水解倾向较小的原因是什么?答:该药物的化学名全称是:1-甲基-4-苯基-4-哌啶甲酸乙酯盐酸盐 该药物结构中虽有酯的结构,但由于苯环的空间位阻的影响,使甲酸乙酯的结构比较稳定,不易水解。



第二节阿片类镇痛药之阳早格格创做一、效率体造阿片类镇痛药又称麻醒性镇痛药( narcotic analgesics ),是一类能与消或者减少痛痛并改变对于痛痛情绪反应的药物.除少量效率强的药物以中,此类药物若使用不当多具备成瘾性,但是用于调理脚段本去不会戴去太大问题.钻研隐现缓性痛痛患者少暂采与阿片类药物治疗时,成瘾的爆收率极矮.表附录12 阿片受体激动后的效率阿片类药物的镇痛效率体造是多仄里的:中周神经有阿片受体;阿片药物可与位于脊髓背角胶状量(第二层)感觉神经元上的阿片受体分离,压造 P 物量的释搁,进而遏止痛痛传进脑内;阿片物量也可效率于大脑战脑搞的痛痛中枢,收挥下止痛痛压造效率.二、阿片类药物的分类阿片类药物有多种分类要领:1. 按化教结构:分为吗啡类战同喹啉类,前者即天然的阿片死物碱(如吗啡、可待果) , 后者主假如罂粟碱,有仄滑肌紧张效率.表附录13 强阿片类药物简表表附录14 强阿片类药物简表2. 按根源该类药物可分为天然阿片类、半合成衍死物 ( 如单氢可待果,二乙酰吗啡 ) 战合成的阿片类镇痛药.合成药物又分为四类:①苯丙吗啡烷类 (phenylpiperidinederivatives) ,如哌替啶、芬太僧等;②吗啡喃类(morphinenans) ,如左吗喃;③苯同吗啡烷类(bengmorphans) ,如喷他佐辛;④二苯甲烷类(diphenylmethanes) ,如好集酮.3. 按受体典型可分为μ、κ、δ受体,该三种受体的分子结构已被决定,并被乐成克隆.从功能上还大概存留ε战δ受体,并大概进一步分为μ 1 、μ 2 、κ 1 、κ 2 、κ 3 战δ 1 、δ 2 等亚型.表 32 为受体激动后的药理效率.4. 按药理效率分,阿片类镇痛药又可分为激动药 ( 吗啡、芬太僧、哌替啶等 ) ,激动一拮抗药 ( 喷他佐辛、纳布啡等 ) ,部分激动药(丁丙诺啡)战拮抗药 (纳洛酮等) .激动—拮抗药又称部分激动药,主要激动κ受体,对于δ受体也有一定激动效率,而对于μ受体则有分歧程度的拮抗效率.由于对于受体效率分歧,那类药物通过κ受体爆收镇痛战呼吸压造效率,有“天花板”效力,很少爆收依好性;通过σ 受体爆收粗神效率战幻觉.根据激动—拮抗程度分歧,纳布啡战布托啡诺主要用做镇痛药,而另一些药如烯丙吗啡主要用做拮抗药.正在临床应用中,已应用杂激动药治疗的患者不克不迭换用混同激动一拮抗药或者部分激动药,可则大概引导戒断反应,而用混同激动—拮抗药或者部分激动药举止治疗的患者可较仄安天换用杂阿片激动药,不会爆收戒断反应.5. 根据阿片类药的镇痛强度,临床分为强阿片药(表附录13) 战强阿片药(表附录14) .强阿片药如可待果、单氢可待果,强阿片药包罗吗啡、芬太僧、哌替啶、舒芬太僧战雷米芬太僧.强阿片药主要用于沉至中度慢缓性痛痛战癌痛的治疗,强阿片类则用于齐身麻醒诱导战保护的辅帮用药以及术后镇痛战中至沉度癌痛、缓性痛的治疗.表附录15 时常使用阿片类药的效率强度战药代教参数表附录1-6 阿片类药物剂量换算表阿片类药的效率强度战药代教本量分歧 ( 表附录15) .表附录16 为临床时常使用的阿片类药物剂量换算表.三、阿片类药物的临床给药道路战要领阿片类药物是暂时已创造镇痛效率最强的药物,而且不“天花板”效力,镇痛效率随剂量的减少而巩固,果此本去不存留所谓最大或者最好剂量.对于个体患者而止,最好剂量由镇痛效率与可耐受不良反应之间的仄稳决断,若判决患者对于阿片类药物仅部分敏感 ( 如部分神经病理性痛痛 ) ,则不该再减少剂量.果此,正在赢得镇痛效率的共时处理阿片类相闭不良反应具备要害意思.( 一 ) 临床药理脂溶性、离子化程度战蛋黑分离率正在决断起效时间、峰时间战效率时间上起主要效率.脂溶性下、分子量小的药物有较下的死物膜渗透性.非离子化药物的脂溶性比离子化药物大 1000 ~ 10000 倍,故非离子化药物的比率愈下,可被弥集进中枢神经系统的药物愈多,起效愈快.蛋黑分离力效率药物的再分集是果为惟有已被分离的药物可弥集透过死物膜,蛋黑分离率下,可用做补偿血浓度落矮的储备量也较多.( 二 ) 终终半衰期直交随分集容积变更并与扫除率相闭.分集容积大,排除半衰期延少,扫除率减少,则排除半衰期支缩.故芬太僧虽扫除率下,但是分集容积大,半衰期仍少.除雷米芬太僧主要由黑细胞战骨骼肌中的非特同性酯酶代开中,其余阿片类药物的代开主要正在肝净中举止,与肝血流相闭.( 三 ) 给药道路无创给药 ( 心服、经皮等 ) 是治疗缓性痛痛、癌痛的尾选给药办法,对于无创要领给药无效以及脚术战脚术后镇痛的患者则采用持绝或者单次静脉给药、持绝或者单次硬膜中给药,也不妨用持绝皮下给药或者临时性肌注给药.为预防或者缩小中周阿片受体激动引导的不良反应,集结收挥中枢镇痛效率,新的给药道路正正在夸大应用.包罗经心腔粘膜、鼻腔粘膜、眼结膜给药等.1) 经心腔粘膜吸支芬太僧 (oral transmucosal fentanyl) 将枸橼酸芬太僧搞成糖块,患者含服时,芬太僧经心腔战食管粘膜吸支直交加进血液循环,仅小部分随唾液加进胃肠,使与胃肠讲阿片受体分离的药物明隐缩小,也落矮了恶心、呕吐战便秘的爆收率.此种给药办法已乐成用于癌痛的突收性痛痛治疗、小女术前用药战小女诊疗性支配.经鼻粘膜战经眼结膜给药共样有预防肝净尾过效力战缩小阿片受体与胃肠讲阿片受体分离的便宜,暂时主要用芬太僧 ( 滴鼻 ) 战舒芬太僧.2) 经皮给药芬太僧脂溶性下,分子量小,镇痛效率强,无局部刺激战皮肤代开,死物利用度下.芬太僧透皮揭剂( 多瑞凶 ) 揭于皮肤后 12 ~ 24h ,血药浓度渐降至稳态并保护 72h .便秘爆收率近矮于心服给药是其主要便宜.该药已广大用于癌痛 ( 提供前提镇痛 ) 战缓性痛痛治疗.3) 患者自控镇痛 (PCA) PCA 是患者感觉痛痛时按压 PCA 开用键,由镇痛泵背体内自动注射设定剂量药物的要领.其特性是医师树立背荷剂量 ( 尽量达到治疗窗浓度 ) 、持绝给药量 ( 保护前提镇痛 ) 、冲打量 ( 统造突收痛或者动做前提镇痛不脚的补充 ) 战锁定时间 ( 预防冲打量尚已收挥效率,患者反复按压开用键引导药物蓄积 ) ,患者按镇痛所需调控镇痛药的注射时机战剂量,是符合于分歧患者、分歧痛痛时间战强度的个体化给药要领,也是国际上通用的术后镇痛给药要领.PCA 分为静脉 PCA(PCIA) 、硬膜中 PCA(PCEA) 、皮下PCA(PCSA) 战中周神经阻滞 PCA(PCNA) .PCIA 采与的主要镇痛药为阿片类药 ( 吗啡、芬太僧、舒芬太僧、阿芬太僧、瑞芬太僧 ) 或者直马多,为预防阿片类药物的恶心、呕吐等不良反应,常加用胃复安、天塞米紧、 5 — H T 、受体拮抗药或者小剂量氟哌啶 (5mg / d 以下 ) ,也可复合非甾体抗炎药以缩小阿片类药物的用量.PCEA 则常采与矮浓度罗哌卡果、布比卡果或者利多卡果等局麻药复合芬太僧、舒芬太僧、吗啡等药物.可加用小剂量可乐定,与局麻药战阿片类药物均有协共效率.( 四 ) 副效率阿片类药的副效率本量是阿片的受体效力.可分为短时间耐受战万古间耐受二大类.镇定、意识朦胧 ( 包罗幻觉 ) 、嗜睡、恶心、呕吐、瘙痒及尿潴留皆是短促反应,数天或者 1 — 2 周后那些症状可消得.最顽固战少期的副效率是便秘,睹于所有强、强阿片类药.耐受性战躯体依好性也是少暂用药后的副效率.阿片耐受性爆收缓缓,各别病人大概果基果突变引导对于吗啡耐受.躯体依好表示为突然停药时出现戒断症状,可通过渐渐减量去预防那种局里.。

阿片类药物的简介一.分类1.按化学结构分类分为吗啡类和异喹啉类,前者即天然的阿片生物碱(如吗啡、可待因),后者主要是提取的罂粟碱,不作用于阿片受体,有平滑肌松弛作用。
2.按来源分类该类药物又可分为天然阿片类、半合成衍生物(如双氢可待因、二乙酰吗啡)和合成的阿片类镇痛药。
合成药物又分为4类:①苯哌啶类(phenylpiperidine derivatives),如哌替啶、芬太尼等;②吗啡烷类(morphinenans),如左吗喃、左啡诺(levorphanol);③苯并吗啡烷类(bengmorphans),如喷他佐辛;④二苯甲烷类(diphenylmethanes),如美沙酮(methadone),右丙氧芬(dextroproxyphene)、镇痛新(pentazocine)3.按受体类型分类可分为μ、κ、δ受体激动剂,该3种受体的分子结构已被确定,并被成功克隆。
然而阿片类受体在中枢神经系统内分布以及对不同阿片受体配型的结合能力存在差异。
阿片受体内源性配体为脑啡肽、强啡肽和内吗啡肽(endomorphine)。
这些五肽物质分别有不同的基因编码,对不同阿片受体的亲和力不同。
脑啡肽对δ受体有较强的选择性,强啡肽对κ受体有较强的选择性,μ受体的内源性配体为内吗啡肽,内吗啡肽在中枢神经系统内与μ受体成镜像分布,其结合力比对δ和κ受体强100倍以上。
而以前所认为的β内啡肽并不是μ受体的内源性配体。
4.按药理作用分类阿片类镇痛药又可分为激动药(吗啡、芬太尼、哌替啶等),激动—拮抗药(喷他佐辛、纳布啡等),部分激动药(丁丙诺啡)和拮抗药(纳洛酮、纳曲酮、去甲纳曲酮等)。
5.根据阿片类药的镇痛强度分类临床分为强阿片药和弱阿片药。
弱阿片药如可待因、双氢可待因,强阿片药包括吗啡、芬太尼、哌替啶、舒芬太尼和瑞芬太尼。
弱阿片药主要用于轻至中度急慢性疼痛和癌痛的治疗,强阿片类则用于全身麻醉诱导和维持的辅助用药以及术后镇痛和中至重度癌痛、慢性痛的治疗。

阿片类镇痛药第一节:概述:疼痛是一种因组织损伤或潜在的组织损伤而产生的痛苦感觉,常伴有不愉快的情绪甚或心血管和呼吸方面的变化。
——它既是机体的一种保护性机制,提醒机体避开或处理伤害,也是临床许多疾」病的常见症状.剧烈疼痛不仅给患者带来痛苦和紧张不安等情绪反应,还可引起机体生理功能紊乱, 甚至诱发休克。
控制疼痛是临床药物治疗的主要目的之躯体痛(Somaticpai n):快痛、慢痛,对机械性、化学性、炎症性、温度性刺激均敏感。
内脏痛(Visceral pai n):对牵张、炎症刺激敏感。
神经痛(Neuropat hi c pai n):多由神经损伤或兴奋性增高引起,呈发作性或持续性,一般镇痛药无效。
躯体疼痛分两类:快痛(剧痛):(Fast pain, acute pain, sharp pai n)尖锐而定位清楚的刺痛,刺激时立即发生,撤除刺激立即消失。
由A S -类纤维传导。
慢痛(钝痛)(chro ni c pa in, sl ow pa in, blunt pain)定位不明确的烧灼痛,发生较慢,持续时间较长。
疼痛除了使患者感受痛苦外,常伴有情绪、心血管和呼吸等方面的变化。
剧痛常引起失眠或其它生理机能的紊乱,甚至引起休克。
由无髓鞘的G类纤维传导。
阿片那与疼痛产生有关的物质:绝大多数情况下,伤害性神经末稍的有效刺激即为化学物质, 包括: 神经递质类(5-HT 、组胺、ACh )、激肽类(缓激肽、赖氨酰缓激肽)、 代谢产物(ATP 、ADP 、H+、K+)、 前列腺素类(PGE2)、 辣椒素(Capsai ci n )。
与疼痛传导有关的神经递质和调质:许多物质参与了痛觉信号的传递和调控过程,包括: 神经肽类:P 物质(SP )、神经激肽(NKA 、NKB ) 经典递质类:Gu )、GA3A 、5 — HT 、NA 腺嘌呤.阿片肽类(opi oi d pept i des )亮氨酸脑啡肽、甲硫氨酸脑啡肽、内啡肽、强啡肽、 内吗啡肽等。


阿片类镇痛药物作者:赵成龙来源:《黄河黄土黄种人》2016年第06期阿片类药物是目前疼痛治疗的主力军。
“阿片”一词广义是指从罂粟的汁液中提取出的药物,包括天然产物吗啡、可待因,二甲基吗啡以及从中提取的许多半合成物质。
生物碱可止痛,鸦片中含有20多种生物碱,其中吗啡的含量最多(9%~17%),因此,吗啡是目前应用最广泛的阿片类镇痛药物。
鸦片战争给我国人民带来了灾难和痛苦,并留下了深深的烙印,这是很多国人恐惧阿片类药物的历史原因,也导致了人们对阿片类药物镇痛治疗的错误认识。
错误认识一:用阿片类药物会成瘾用阿片类药物镇痛,成瘾性的发生率与药物的给药方式有关。
静脉注射大量止痛药物,会使血液中药物浓度突然增高,脑内药物浓度也明显增高,超过所需要的止痛药浓度,易成瘾。
在慢性疼痛中,采用阿片类药物的控缓释制剂,药物在胃肠道内缓慢释放,使血液中药物浓度在一定程度上保持恒定,成瘾现象极其罕见。
在临床上常用的阿片类药物包括吗啡控释片、羟考酮控释片、芬太尼透皮贴剂、盐酸吗啡注射液、盐酸吗啡片等。
长期的临床实践证明,以止痛治疗为目的,阿片类药物在常规剂量、规范化使用情况下,疼痛患者出现成瘾的现象极为罕见。
国外大型临床实验证实成瘾的患者只占0.029%,也就是说成瘾性发生率不足万分之三。
错误认识二:用阿片类药物意味着“临终期”将至目前,由于众所周知的原因,癌症的发病率越来越高,而且至少有50%的中、晚期癌症患者伴有不同程度的疼痛。
随着肿瘤治疗技术的进步,有一部分患者可取得较好的治疗效果,而经过有效的抗肿瘤治疗后,阿片类药物是可以减量甚至停用的。
而有些患者认为自己得了癌症,肯定活不成了,“临终期”将至,于是任由疼痛持续存在,坚持不服阿片类药物。
久而久之,导致患者生存欲望降低、治疗依从性差、机体免疫力下降等恶性循环,这样就缩短了生存期。
错误认识三:疼时再服阿片类药物部分患者担心服用阿片类药物会成瘾或出现其他身体不适,内心深处抵触服药。


第七章阿片样镇痛药*阿片(opium)即鸦片的简介(来源、用途)阿片是罂粟科植物罂粟的未成熟蒴果被划破后流出的白色液汁,干燥后呈棕黑色膏状物。
→阿片内含多种复杂成分,其中的生物碱具有药理活性。
阿片中分离到20多种生物碱,其中的主要成分是吗啡,其它成分尚有可待因(Codeine)、蒂巴因(Thebaine)等。
吗啡是用于临床的镇痛药;可待因镇痛作用为Morphine的1/10,主要用作镇咳药;蒂巴因为半合成镇痛药(阿片样激动剂埃托啡(eterphine)、阿片样拮抗剂纳络酮(nalotone)等)的合成原料。
吗啡于1806年从阿片中分离提取出来,1923年确定了化学结构,1952年完成了合成工作。
*吗啡的药理活性:吗啡除具有强镇痛活性(显著减轻或消除疼痛)、镇静和欣快作用外,还有严重的副作用。
例如治疗剂量时呼吸抑制、血压降低、恶心呕吐、大小便困难、嗜睡等,最为严重的不良反应是吗啡反复使用,易产生耐受性、成瘾性,一旦停药即出现戒断症状,危害极大。
寻找成瘾性小、不良反应少的理想镇痛药成为研发新镇痛药的目标。
*在研究构效关系、开发新镇痛药方面所做的工作:1、对吗啡进行结构修饰——半合成镇痛药。
2、简化吗啡结构——合成镇痛药和吗啡拮抗剂;——具有阿片样激动/拮抗作用成瘾小的镇痛药、高效镇痛药。
(近年的发展)*阿片受体的外源性配体、内源性镇痛物质、阿片受体阿片样镇痛药作用机理的研究认为:阿片样镇痛药(Opioid Agents)通过与体内高度特异性受体部位结合后产生药理活性。
阿片样镇痛药是阿片受体的外源性配体,与阿片受体相互作用产生减轻剧烈锐痛或钝痛等药理活性。
阿片受体的外源性配体及内源性镇痛物质阿片样镇痛药(阿片样激动剂、拮抗剂和激动/拮抗剂)是阿片受体的外源性配体;脑啡肽、内啡肽、强啡肽等是内源性镇痛物质。
阿片受体与其内源性配体相互作用,除调节疼痛感觉外,还有重要的生理功能。
(继1975年发现内源性具有吗啡样镇痛活性的脑啡肽之后,又发现了内啡肽、强啡肽等内源性镇痛物质。
)现代科学技术证实:人类的脑内、脊髓组织中、外周神经系统存在阿片受体。
体内阿片受体通常被认为主要分为μ(mu)、κ(kappa)、δ(delta)三种。
*阿片样镇痛药的作用:药物通过模拟内源性阿片样肽类与阿片样物质受体(Opioic receptor)(简称阿片受体)相互作用,通过影响局部神经元及体内的疼痛调节回路,导致产生镇痛及其他治疗效用和副作用(导致产生多种药理作用)。
*阿片样镇痛药的分类:阿片样镇痛药按其来源可分为四类:阿片生物碱类;半合成镇痛药;合成镇痛药和内源性阿片样肽类。
阿片样镇痛药按其作用机理可分为三类:阿片受体激动剂;混合的激动-拮抗剂(阿片受体部分激动剂)和阿片受体拮抗剂。
*阿片样镇痛药受国家颁布的《麻醉药物管理条例》管理。
第一节吗啡及相关的阿片样激动剂吗啡(Morphine)、哌替啶(Pethidine)为μ受体激动剂,强啡肽为κ受体激动剂,脑啡肽为δ受体激动剂,喷他佐辛(Pentazocine)、丁丙诺啡(Buprenorphine)为混合的激动-拮抗剂(Mixed Agonist-Antagonist),纳洛酮(Naloxone)为阿片受体拮抗剂。
一、阿片生物碱类(一)盐酸吗啡(Morphine Hydrochloride)(μ阿片受体激动剂)1.吗啡的化学结构药用吗啡从阿片中提取制备,具有左旋光性,临床用其盐酸盐或硫酸盐。
吗啡的镇痛活性与其立体结构严格相关,仅(-)-吗啡有活性。
天然的吗啡具有左旋光性,左旋吗啡((-)-Morphine)是由5个环稠合而成的刚性结构。
A、B和C环构成部分氢化的菲环,C和D环构成部分氢化的异喹啉环,分子中有5个手性中心(5R,6S,9R,13S,14R),B/C环呈顺式,C/D环呈反式,C/E环呈顺式,环D呈椅式构象,环C呈半船式构象,环A以直立键连接在环D(哌啶环)的4位上。
(-)-吗啡的构象呈三维的"T"形,环A,B 和E构成"T"型的垂直部分,环C,D为其水平部分。
吗啡结构中3位有酚羟基,呈弱酸性; 17位的叔氮原子呈碱性,因此能与酸或强碱生成稳定的盐使水溶性增加。
临床上常用其盐酸盐。
右旋吗啡无镇痛及其他生理活性,自然界中不存在,只能人工合成。
2.吗啡的性质:白色针状结晶或结晶性粉末,无臭味苦,能溶于水,极易溶于沸水,略溶于乙醇,不溶于氯仿、乙醚。
具有左旋光性。
稳定性(1)3位酚羟基的存在,使吗啡及其盐的水溶液不稳定,放置过程中,受光催化易被空气中的氧气氧化,变色变深。
生成毒性大的双吗啡(Dimorphine)或称伪吗啡(Pseudomorphine),氧化反应机理为自由基反应。
产物还有N-氧化吗啡和微量的甲胺。
吗啡的稳定性受pH和温度影响,pH4最稳定;中性和碱性条件下,受紫外线照射或重金属离子(如铁离子)的催化可加速其氧化反应。
(2)吗啡与盐酸或磷酸加热反应,经分子重排生成具有邻二酚结构的阿扑吗啡(Apomorphine),更易被氧化。
在碱性(碳酸氢钠)条件下阿扑吗啡(Apomorphine)水溶液被碘试液氧化,生成邻醌化合物。
该氧化产物在有水和乙醚存在时,水层呈绿色,醚层呈宝石红色,中国药典用此反应对盐酸吗啡中的杂质阿扑吗啡作限量检查。
(3)吗啡在酸性条件下与亚硝酸钠反应,生成2-亚硝基吗啡,加入氨水至碱性时显黄棕色。
可待因无此反应,可以以此反应对可待因中的杂质吗啡作限量检查。
3.吗啡的体内代谢吗啡口服,因肝脏的首过效应是吗啡的生物利用度仅为25%。
主要代谢反应为:吗啡3位葡萄糖醛酸轭合物几乎没有活性吗啡6位葡萄糖醛酸轭合物为活性代谢物次要代谢反应为:吗啡N-脱甲基生成去甲基吗啡。
4.吗啡的用途吗啡为μ阿片受体强激动剂,镇痛作用强,具有镇静作用。
不良反应包括连续使用成瘾可产生耐受和依赖,呼吸抑制等,滥用危害极大。
需按国家颁布的《麻醉药品管理条例》管理(二)磷酸可待因 (Codeine Phosphate)→←↑——1.可待因的性质:磷酸可待因具左旋光性,为白色细微针状结晶或结晶性粉末。
可待因在氨试液中有一定的溶解性,磷酸可待因水溶液可溶于氨试液而不会出现沉淀。
可待因在阿片中含量较低,主要以吗啡为原料经甲基化反应制备,产品中可能引入吗啡,药典采用吗啡与亚硝酸反应后,在氨碱性下显棕黄色(可待因无此反应),进行限量检查。
可待因分子中无游离酚羟基,性质较吗啡稳定,但遇光仍易变质,需避光保存。
2.可待因的体内代谢代谢反应为:可待因N-脱甲基生成去甲基可待因。
3.可待因的用途可待因为弱μ激动剂,镇痛作用为吗啡的1/10,用于中等程度的止痛,主要用作中枢性镇咳药。
耐受性、成瘾性和呼吸抑制等副作用均小于吗啡,通常药用其磷酸盐。
二、早期对吗啡的结构修饰对吗啡进行结构修饰——半合成镇痛药为增强镇痛作用,降低毒副作用,对吗啡结构进行修饰,得到半合成镇痛药即吗啡的衍生物。
吗啡衍生物结构特点:保留吗啡的五环基本结构,化学合成困难。
吗啡的结构改造:1.3位酚羟基烷基化(3位酚羟基的氢被烷基取代),镇痛活性降低。
酚羟基的氢由甲基取代——可待因(甲基吗啡),镇痛为吗啡的1/10。
酚羟基的氢由乙基取代——狄奥宁(乙基吗啡),镇痛为吗啡的1/10。
可待因 (Codeine) 海洛因2.3位酚羟基和6位醇羟基的酯化二羟基中的H被乙酰化(吗啡分子中的两个羟基酯化为二乙酸酯)——海洛因(双乙酰吗啡)毒品!!酯化后亲酯性增强,易透过血脑屏障到达中枢。
海洛因代谢物6-乙酰吗啡对μ受体的激动作用强于吗啡,欣快感更强,成瘾性、耐受性和依赖性高于吗啡,危害极大。
3.C7-8双键氢化, C6位醇羟基氧化成酮临床用于镇痛的药物:C7-8双键氢化,C6位羟基氧化成酮,生成氢吗啡酮,副作用、成瘾性较小,镇痛效用约为吗啡的8——10倍。
(用于临床)在氢吗啡酮基础上所作的工作:C7-8双键氢化,C6位羟基氧化成酮,C14上引入羟基,生成羟吗啡酮,镇痛效用约为吗啡的10倍,副作用也大。
(用于临床,镇痛作用为吗啡的10倍,副作用增大。
)C7-8双键氢化,C6位羟基氧化成酮,C14上引入羟基,C3位羟基甲基化,生成羟考酮。
(用于临床)C7-8双键氢化,C6位羟基氧化成酮,C3位羟基甲基化,生成氢可酮。
(用于临床,镇痛作用低于吗啡。
)4.17位N上甲基的改变(由其它烷基、链烷基、芳烃基取代)(1)由苯乙基替代烃——→苯乙基吗啡,镇痛作用为吗啡的6倍。
(2)由稀丙基替代——→稀丙吗啡,(Nalorphine)(纳洛芬),μ受体拮抗剂,对抗吗啡中枢抑制作用,是吗啡类镇痛药中毒的解毒药。
临床常用其氢溴酸盐。
苯乙基吗啡纳洛芬(烯丙吗啡)小结: C3位游离酚羟基的存在对吗啡的镇痛活性是重要的;C6位醇羟基成酮、C7-8位双键被氢化、N14位引入羟基,一般均使活性增强,毒性也增强;N17-甲基若被稀丙基取代,可得到拮抗剂。
(3)C7-8双键移位、C-14氢由羟基取代、N-甲基改变为稀丙基——纳洛酮纳洛酮(Naloxone)为阿片受体纯拮抗剂,可有效的拮抗具有激动活性或混合的激动-拮抗活性的阿片样镇痛药的作用,临床用于此类药物过量时引起的呼吸抑制的解救。
常药用其盐酸盐氢吗啡酮纳洛酮(C7-8位单键、C6位酮基,区别于烯丙吗啡的C7-8位双键)三、其他的μ受体激动剂(合成镇痛药)对吗啡的结构进行简化发展了合成镇痛药。
合成镇痛药按化学结构类型可分为五类:吗啡喃类,苯吗喃类,哌啶类,苯基丙胺类(氨基酮类),氨基四氢萘类等。
(一) 吗啡喃类吗啡喃结构————N-甲基吗啡喃结构吗啡喃类合成镇痛药也称吗啡烃类,吗啡化学结构中去掉C4,C5位之间的醚键,C3和C17位的取代基为氢,C7、C8位双键氢化。
吗啡喃,无镇痛活性。
其立体结构与吗啡相同。
17-甲基吗啡喃-3-醇酒石酸盐二水合物的左旋体称为酒石酸左啡诺(Levorphanol Tartrate)(酒石酸那洛啡尔、左吗喃)。
N-甲基吗啡喃的3位引入-OH后镇痛作用增强,其左旋体为左啡诺,镇痛时间较长,镇痛作用为Morphine的6倍,是强μ受体激动剂。
17-(环丁烷甲基)-吗啡喃-3,14-二醇酒石酸盐称为酒石酸布托啡诺(Butorphanol Tartrate),为μ受体拮抗剂、κ受体激动剂,这种新发展的混合型激动-拮抗剂(Mixied Agonist-Antagonists)用作镇痛药,成瘾性小。
(二)苯吗喃类(苯并吗啡烷类)苯吗喃结构:与吗啡结构相比,去掉E、C两个环,C环开裂后需在原处保留小的烃基作为C环残基,其构象与吗啡相似。
此类药中用于临床的喷他佐辛(Pentazocine),又名镇痛新,为阿片样激动-拮抗剂,它是κ受体激动剂、μ受体弱拮抗剂,镇痛作用弱于吗啡(约为其1/10),成瘾性小。