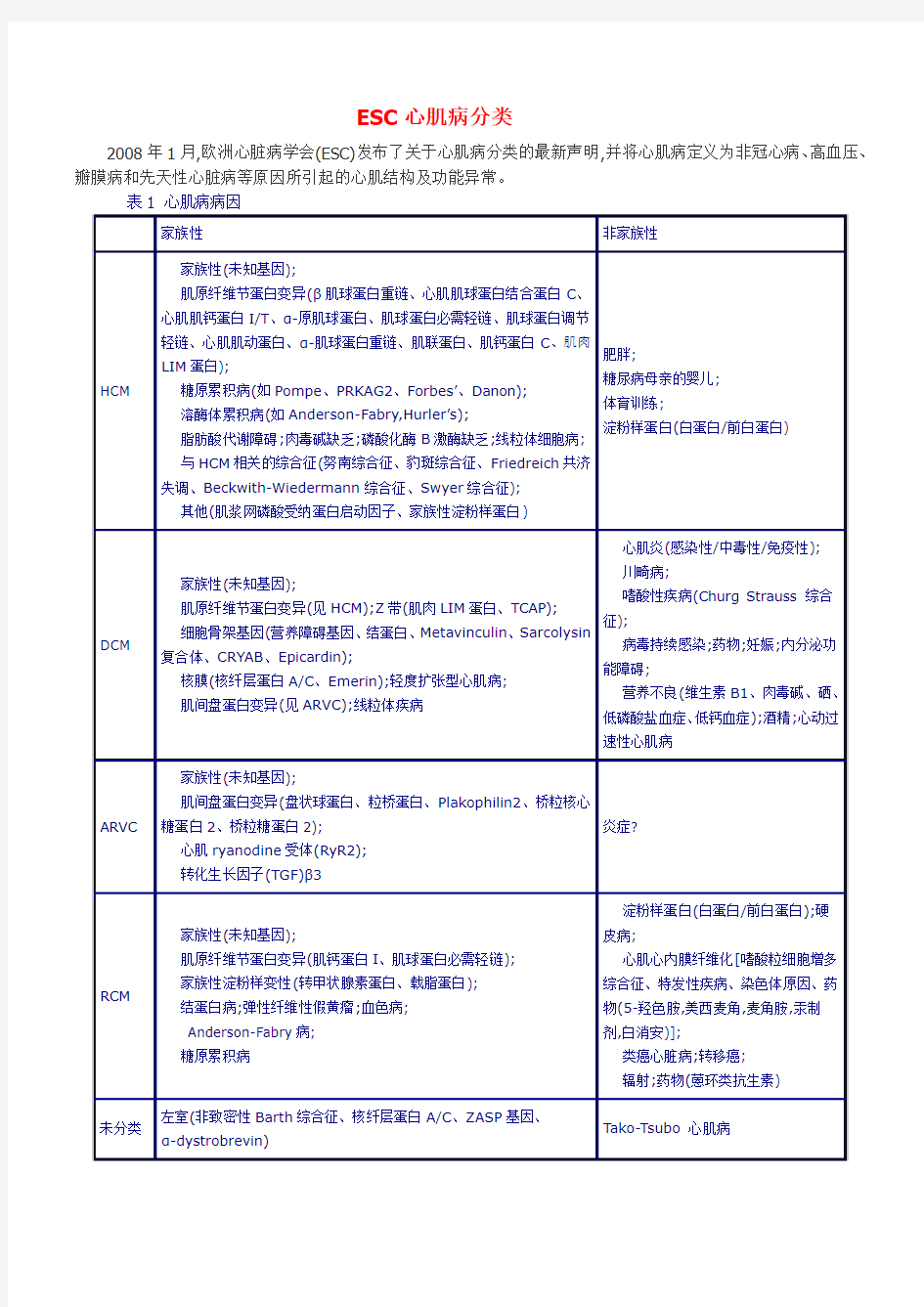

ESC心肌病分类
2008年1月,欧洲心脏病学会(ESC)发布了关于心肌病分类的最新声明,并将心肌病定义为非冠心病、高血压、瓣膜病和先天性心脏病等原因所引起的心肌结构及功能异常。
图1 心肌病分类
新分类四大特色
1. 新分类建立于疾病特殊形态及功能表型之上,而非建立于病理生理机制之上,因此更适于临床应用。
2. 新分类将心肌病进一步划分为家族性和非家族性,注重心肌病的遗传决定因素,并以此指导诊断试验。
3. 不再对原发性和继发性心肌病进行区分。
4. 诊断重心从以排除诊断为主转向寻找积极的、有逻辑性的诊断指标。
心肌病根据形态学特异性和不同功能表现分类,同时又可分为家族性和非家族性。
肥厚型心肌病(HCM)
定义:无导致心肌异常的负荷因素(高血压、瓣膜病)而发生的心室壁增厚或质量增加。
传统的HCM定义还规定不应是淀粉样变性及糖原累积病等系统性疾病而导致的心肌肥厚。目的是为了区分肌细胞肥大或间质侵润及细胞内代谢产物积累所致的室壁增厚。然而在临床实践中,通过超声心动图和磁共振成像(MRI)等无创手段通常不能鉴别上述两种病症;而由于心肌病理的复杂性,心肌活检往往于患者死后才能得以进行。因此本指南对HCM定义进行了简化。
非高血压和瓣膜病引起的左室肥厚发生率约为1/500,多为家族性常染色体显性遗传,由编码心肌肌原纤维节不同蛋白的基因变异所致,且多表现为不对称性心肌肥厚(尤其是室间隔部位)以及肌细胞排列紊乱。左室容积常减少,左室缩短分数高于正常。少数(≤10%)患者可进展为左室扩张和收缩功能障碍。
高强度体育锻炼可引起左室形态发生生理性改变,但心肌厚度极少与HCM患者的病理表型相似(男性运动员<2%)。
扩张型心肌病(DCM)
定义:无引起整体收缩功能障碍的异常负荷因素(高血压、瓣膜病)或冠脉疾病而发生的左室扩张合并左室收缩功能障碍性疾病,伴或不伴右室扩张和功能障碍。
25%的西方患者有家族史,且多为常染色体显性遗传。有家族性早发心源性死亡、传导系统疾病或骨骼肌疾病者应高度怀疑家族性DCM。
心脏感染和炎症的晚期可发生DCM,但有别于急性心肌炎(左室大小常在正常范围),炎症性DCM定义为合并有慢性炎症细胞的左室扩张和射血分数降低。因此,组织学和(或)免疫细胞化学检测是不可缺少的确诊途径。
轻度扩张型充血性心肌病(MDCM)用于描述既无限制性血流动力学障碍,也无明显左室扩张(超过正常上限但小于10%~15%)的严重左室收缩功能障碍的晚期心衰。50%以上的患者具有DCM家族史,临床表现及预后与典型DCM患者相似。
DCM的另一种形式是围产期心肌病(PPCM),表现为妊娠最后1个月或分娩后5个月内出现心衰症状,多见于30岁以上的妇女,与妊娠高血压、双胎妊娠和应用宫缩抑制剂治疗相关。
限制型心肌病(RCM)
定义:在收缩容积正常或降低(单/双心室)、舒张容积正常或降低以及室壁厚度正常的情况下发生的限制性左室生理学异常。
限制性左室生理学异常的特点为由心肌僵硬度增加所致的左室充盈状态,表现为心室压力显著升高而心室容积仅轻度增加。引起RCM的病因包括特发性、家族性和全身系统性疾病。家族性RCM通常为常染色体显性遗传。
损害收缩功能的心内膜病理变化(纤维化、纤维弹性组织增生和血栓)也可导致限制性心室生理学异常,根据嗜酸粒细胞是否增多可进一步将其分为2个亚组:伴嗜酸粒细胞增多的心内膜心肌病(现归类于嗜酸粒细胞增多综合征)和无嗜酸粒细胞增多的心内膜心肌病[如心肌心内膜纤维化(EMF)]。
致心律失常性右室心肌病(ARVC)
定义:右室功能障碍(局部或整体),伴或不伴左室疾病,同时有组织学证据和(或)符合相应标准的心电图异常表现。
ARVC组织学表现为右室心肌被脂肪和纤维组织逐步取代。病变主要累及右室前壁漏斗部、心尖部及后下壁,三者构成“发育不良三角”。尽管ARVC较为罕见,估计发病率为1/5000,但在欧洲某些地区,ARVC是青年猝死
的常见原因。多数ARVC为基因编码plakophilin-2和其他心肌细胞桥粒蛋白变异的常染色体显性遗传,但也有一些病例确认为常染色体隐性遗传。
未分类心肌病
左室致密化不全(LVNC)
LVNC特点为左室具有明显的肌小梁和深部小梁间隐窝。室壁常增厚,心外膜致密变薄而心内膜增厚。一些患者可伴有左室扩张、收缩功能障碍。
目前尚不能明确LVNC是否为一种单独的心肌病。LVNC可单独出现或与先天性心脏疾病如Ebstein畸形或紫绀型心脏病以及神经肌肉疾病联合出现。LVNC通常为家族性,至少25%无症状亲属有不同程度的超声心动图异常。
Tako-Tsubo心肌病
暂时性左室心尖球形综合征或Tako-Tsubo心肌病的特点为短暂的左室心尖和(或)心室中段收缩功能障碍,冠脉造影无阻塞性冠脉疾病。患者可表现突发“心绞痛样”胸痛、广泛T波倒置甚至ST段抬高,以及心肌酶轻度升高。多数报道病例为绝经后女性,通常于症状发生前有情绪激动或生理应激。大多数患者去甲肾上腺素浓度升高。左室功能通常在数天或几周后恢复正常,罕有复发者。
2008年欧洲心肌病分类共识解读
作者:刘文玲日期:2008-4-3刘文玲北京大学人民医院
自1995年世界卫生组织(WHO)/国际心脏病学会联合会(ISFC)心肌病分类出台以来,心肌病的相关研究取得了显著进展,特别是心肌病分子遗传学领域取得了突破性进展,一些心肌病的病因已经明确,并发现了新的心肌病类型。2006年美国心脏病协会(AHA)发表了“现代心肌病定义和分类”,今年伊始欧洲心脏病学学会(ESC)发表了“心肌病分类共识”,ESC的分类与AHA的分类有很大不同。
■心肌病分子机制研究的突破性进展
心肌病领域最大进展为心脏病分子生物学的突破。近年来研究发现,1/3左右的扩张型心肌病为家族遗传性疾病,常伴有骨骼肌和神经肌肉病变。家族性扩张型心肌病(无论是常染色体遗传还是X连锁遗传)基因缺陷为编码蛋白的基因突变如营养障碍基因、糖蛋白、肌聚多糖和destroglycans。由于所有细胞骨架蛋白均与肌肉收缩力的传递有关,因此,家族性扩张型心肌病被认为是一种细胞骨架疾病。已经证明肥厚型心肌病是一种常染色体显性遗传的家族遗传性疾病,为肌力产生障碍的原发性肌原纤维疾病,由于编码收缩蛋白的基因缺陷所致如β肌球蛋白重链基因、肌球蛋白连接蛋白C、atropomyosin和肌钙蛋白T、肌钙蛋白I。异常蛋白造成舒张期肌丝的松弛受损。
致心律失常性右室心肌病(ARVC),一种常染色体显性或隐性疾病,为细胞链接性疾病,即确保心肌细胞间机械性连接的桥粒蛋白发生异常。已经发现编码桥粒斑蛋白、盘状球蛋白、plakophilin、桥粒核心糖蛋白和桥粒糖蛋白的基因突变。右心室的变薄、扩张为桥粒断裂、细胞损伤和纤维脂肪组织替代的结果。
长QT综合征和短QT综合征为钾通道病,Brugada综合征和Lenegre病为钠通道病(SNC5A)。儿茶酚胺依赖性室性心动过速为肉桂碱受体突变,肉桂碱受体控制自肌浆网中的释放。除Lenegre病为累及特殊传导系统的心肌病外,所有这些综合征心脏结构均正常,易发生细胞膜电不稳定。这种改变仅表现在心电图上:分别表现为QT间期延长或缩短、ST 段抬高或劳力诱发的室性心动过速。分子水平的病理基质为编码氨基酸的核酸序列发生了突变,简单的错义突变也会导致蛋白质的改变。
心肌病可以仅表现为电紊乱而无结构异常,纳入了这一概念,这类原发性电紊乱性疾病都应归属为心肌病的范畴,这样我们就可以更多地通过心电图而不是超声心动图和心脏核磁来诊断心肌病。因此,原发性遗传性心肌病可以分为两类:①结构异常性心肌病(扩张型、肥厚型、限制型和致心律失常性),即细胞骨架、肌节、细胞链接病变的心肌病;②无结构异常的心肌病(短QT和长 QT综合征、Brugada综合征和儿茶酚胺依赖性室性心动过速),即通道病。
关于炎症性心肌病,应用原位杂交和多聚酶链反应(PCR)证明肠病毒和腺病毒均是亲心脏病毒。肠病毒性心肌炎导致的扩张型心肌病,分子机制为病毒蛋白酶2A对营养障碍基因复合物的溶解作用导致的后果。
■1995年WHO/ISFC心肌病的定义和分类的不足
1995年WHO/ISFC心肌病的定义和分类将当时新发现的心肌病即ARVC和限制性心肌病等心肌病纳入了其中,但致密化不良性心肌病被分到未分类型心肌病中。1995年分类中继发性心肌病即特异性心肌病的概念过于宽泛,以至于将慢性缺血性、瓣膜性和高血压病均纳入其中。随着病因学的研究进展,原来一些“病因不明的心肌疾病”成为“伴有心脏功能障碍的心肌疾病”。心脏功能障碍包括心脏机械性障碍和心电障碍,随着心脏病分子生物学的进展,原发性电紊乱性疾病应该有所归属。
心肌疾病界定为具有心功能障碍,这就扩展了心肌病的概念。传统上,心肌细胞的功能异常指收缩功能异常,如扩张型心肌病伴有泵功能衰竭,而肥厚型心肌病甚至收缩功能增强,限制型心肌病伴有舒张期松弛受损。而且,心肌功能异常可以是单纯电异常,可以有结构异常如心肌炎和ARVC,也可以无结构异常,如离子通道病无任何收缩功能的损害。
■2006年AHA对心肌病定义和分类的拓展
2006年AHA根据上述新观念出台了新的心肌病定义和分类(图1)。如果心脏功能异常可以是机械性的也可以是电的,那么心肌病不必一定要存在心脏扩张或肥厚。其病因可以为遗传性和获得性。因此,2006年AHA新的定义为“心肌病为一组临床表现为多种多样的心肌疾病,具有结构异常和(或)电异常,由各种原因通常是遗传原因造成,常表现为心室异常肥厚或扩张,但也可以正常”。此分类仍然沿用了原发性和继发性的分类,原发性心肌病指“仅限于心肌或主要累及心肌的疾病”,继发性心肌病指“心肌病变是全身性疾病的一部分(多器官受损)”。原发性心肌病分为三种类型(遗传性、获得性和混合性),将心脏结构正常的原发性电紊乱(离子通道病)和Lenegre病也归入心肌病。摒弃了未分类型心肌病。将心肌病分为家族性/遗传性和非家族性/非遗传性心肌病,有利于筛查基因突变和分析。因此,心肌病的概念中纳入了一大类遗传性心肌病,不仅包括了先前发现的有明显形态学异常的心脏病,还包括了新近发现的表现为原发性心律失常,而无结构改变的疾病。而在此之前,这类疾病一般划分在心律失常的范畴。不断有证据表明,这些“原发性电紊乱”心肌病常常与“传统的结构性”心肌病交错在一起。AHA 工作组建议,除了心肌病的临床表型分类外,按照这种分类方法涵盖了那些遗传缺陷性心肌病包括编码肌小节、细胞骨架、桥粒或离子通道蛋白的基因突变的疾病。
■2008 ESC 共识的局限性
ESC工作组就1995年WHO/ISFC 的心肌病分类的修订发表了相关声明(图2)。在该定义中,明确宣布心肌病为非冠状动脉疾病、高血压、瓣膜病和先天性心脏缺陷导致的心肌结构和功能异常的心肌疾病。
该共识接受并重申AHA 将心肌病分为家族性/遗传性心肌病和非家族性/非遗传性的分类,摒弃传统的原发性和继发性(特异性)心肌病的分类,因为这种分类可能造成一些误解,错误地认为原发性就是自发性、继发性就是已知病因的心肌病,否认单纯电紊乱为心肌病,从而将离子通道病和传导系统疾病排除在心肌病范畴之外。基本上按照形态功能表现分为五种类型心肌病(肥厚型、扩张型、致心律失常性、限制型和未分类型),包括家族性或非家族性,无论是否单纯表现为心脏受累。这种分类无疑是将复杂病情难题简单化,但是,不能完全回答临床上出现的问题。当去掉特异性心肌病后如缺血、高血压和瓣膜病后,这些疾病的归属应该受到关注。由于AHA在2006年的分类中首先去掉了特异性心肌病,但心肌炎却划分到心肌病的范围内,这不能令人信服。AHA 声明摒弃了未分类心肌病,然而,ESC 声明仍然认为有些类型如致密化不全和Tako Tsubo 心肌病应该有一席之地。ESC将只有结构异常者归类为心肌病,未考虑疾病表型的自然病程中一种疾病可能发展为另一种疾病,如有研究者发现Brugada综合征可能是致心律失常性右室心肌病(ARVC)的早期表现。将电紊乱性疾病排除在心肌病的范畴,有悖心肌病概念中的“功能异常”。
■结论
AHA 和 ESC的声明对心肌病均有很大贡献,使这一复杂领域更加明朗。由离子通道病导致的电紊乱性心肌病仍然受到很大的关注。由于理论上电紊乱可能从心室扩展到心房,家族性心房颤动可能也应属于遗传性心肌病的范围。临床上,疾病表型十分复杂,这种分类可能造成疾病之间的重叠。根据基因学和蛋白组学心血管方面的突破性进展,世界各地专家认为,必须修订1995 WHO/ISFC的分类以达成共识。
中国扩张型心肌病诊断和治疗指南要点 扩张型心肌病(DCM)是一类异质性心肌病,是心力衰竭和猝死的常见疾病之一。以往对DCM局限于心力衰竭和心律失常的治疗,2018年发表的中国扩张型心肌病诊断和治疗指南(简称指南)提出病因诊断及其检测方法,针对免疫学病因早期治疗,将基础研究转化到临床研究,对DCM诊断与治疗展现出新的理念与方法。 1.扩张型心肌病病因诊断的创新与临床实践 指南将扩张型心肌病分为原发性和继发性2类,原发性DCM包括家族性DCM、获得性DCM和特发性DCM。家族性DCM推荐临床开展基因检测,明确病因诊断。在获得性DCM类型中提出免疫性DCM,以抗心肌抗体(anti-heart antibodies,AHA)作为免疫学标记物。1985年Schultheiss等发现扩张型心肌病(DCM)患者抗心肌线粒体腺嘌呤核苷异位酶(ANT)自身抗体干扰心肌细胞能量代谢导致心肌病,2011年笔者发现DCM患者抗L型钙通道抗体(抗L-CaC抗体)可以引起室性心律失常和猝死,并阐明其作用机制,并且建立抗心肌抗体的检测方法。 2014年浦介麟等临床研究证实DCM组(?? = 732)和对照组(?? = 834)随访52个月,DCM患者抗L-CaC抗体阳性组总死亡、全因死亡和
猝死均显著高于对照组;2062例慢性心力衰竭(CHF)与824例对照组人群随访36个月,在CHF患者379例死亡中,猝死率DCM组40.37%和ICM组39.07%,DCM和ICM组抗β1肾上腺素受体(β1AR)抗体阳性患者的猝死率显著高于对照组。中国多项临床观察性研究证实抗L-CaC 抗体和抗β1AR抗体阳性具有CHF死亡和DCM猝死的独立预测价值。然而,国内外资料显示AHA在41%~85%的特发性DCM、60%的FDCM 和46-60%的PPCM患者中被检出阳性,该指南推荐对于心脏扩大非缺血性心衰患者,常规检测抗心肌抗体,提供DCM免疫诊断、指导选择针对性治疗和预测DCM猝死和死亡风险。 2. 扩张型心肌病的早期病因治疗:免疫学治疗 2014年中国报道767例DCM随访52个月死亡率为42.24%。为了降低DCM死亡率,需要针对DCM的早期诊断和早期干预。在DCM早期阶段,该指南积极推荐针对DCM病因治疗包括免疫学治疗和尽早应用神经激素拮抗剂治疗,可减少心肌损伤和延缓病变发展。该指南创新性提出扩张型心肌病免疫学治疗的方法。 2.1阻止抗体致病作用的治疗:指南推荐了针对抗β1AR抗体和抗 L-CaC抗体阳性的DCM早期患者应用β受体阻滞剂和地尔硫卓治疗。国内外多中心随机临床试验(J-CHF、MDC、DiDi、ISDDC试验)证实β受
肥厚型心肌病诊断治疗指南 肥厚型心肌病(HCM)是一种原因不明的心肌疾病,特征为心室壁呈不对称性肥厚,常侵及室间隔,心室内腔变小,左心室血液充盈受阻,左心室舒张期顺应性下降。根据左心室流出道有无梗阻分为梗阻性及非梗阻性肥厚型心肌病,可能与遗传等有关。肥厚型心肌病有猝死风险,是运动性猝死的原因之一。 病因 肥厚型心肌病是常染色体显性遗传性疾病,60%~70%为家族性,30%~40%为散发性,家族性病例和散发病例、儿童病例和成年病例具有同样的致病基因突变。目前已证实,至少14个基因突变与肥厚型心肌病的发病有关,其中有10种是编码肌小节结构蛋白的基因,绝大部分突变位于这些基因。 临床表现 1.以青壮年多见、常有家族史。
2.可以无症状,也可以有心悸、劳力性呼吸困难、心前区闷痛、易疲劳、晕厥甚至猝死,晚期出现左心衰的表现。 3.梗阻性肥厚型心肌患者胸骨左缘可出现粗糙的收缩中晚期喷射性杂音,可伴震颤,应用洋地黄制剂、硝酸甘油、静点异丙肾上腺素及Valsalva动作后杂音增强,反之应用β受体阻滞剂、去甲肾上腺素、下蹲时杂音减弱。有些病人闻及S3及S4心音及心尖区相对性二尖瓣关闭不全的收缩期杂音。 检查 1.超声心动图 对HCM诊断有重要意义:①室间隔肥厚与左室游离壁厚度之比>1.5cm;②二尖瓣前叶收缩期向前移动及主动脉收缩中期关闭现象;③心室腔小;④左室流出道狭窄<2.0cm;⑤左室流出道血流速度加快;⑥休息时收缩期左室心尖部心腔与流出道压力阶差>30mmHg,则认为存在左室流出道梗阻。对称性左室肥厚时室间隔与左室游离壁一致。 2.心电图
左心室或双室肥厚及ST-T改变,深而倒置的T波、有时有异常Q波。房室传导阻滞和束支传导阻滞。还可以发现其他心律失常如房颤、早博等。 3.X线检查 X线检查没有明显的特点,可能见到左房、左心室增大,也可能在正常范围。晚期可见右室增大和肺淤血表现。 4.心脏磁共振(MRI) 其敏感性高于超声心动图,但费用较高,对于诊断特殊部位的肥厚和不典型的肥厚最为灵敏。还可以发现心肌纤维化组织。 5.心内膜下心肌活检 免疫性荧光可发现肥厚心肌内儿茶酚胺含量增高,组织学可见心肌排列紊乱和肥大的心肌细胞。 诊断 超声心动图提示左心室壁或(和)室间隔厚度≥15mm,排除了其他引起心肌肥厚的原因如高血压病、风湿性心脏病二尖瓣病、先天性心脏病(房间隔、室间隔缺损)及代谢性
2015 年三基三严培训:心肌病的分类试题 姓名:分数: 一.填空题:(每题 5 分,共16*5=80 分) 1. 心肌病的分类:(扩张型心肌病)(肥厚型心肌病)(限制型心肌病)(致 心律失常性右室发育不全心肌病)(未分化的心肌病)(特异性心肌病) 2. 扩张型心肌病(DCM ):是以(左心室)或(双心室)扩张和收缩功能受损为体 征。 3. 特异性心肌病是指与特异性心脏病或系统性疾病有关的心肌疾病,常见的有(缺血性 心肌病)(瓣膜性心肌病)(高血压性心肌病)等。 4. 扩张性心肌病占心衰人数的(10-15%),死亡率高,10 年生存率(10-30% )。出现 心衰症状后, 5 年生存率(40%) 5. 扩心病常见的症状是(室性心律失常)(猝死)。 二、问答题(共10*2=20 分) 1.扩心病的的治疗原则:强心、利尿、ACEI 等药物治疗外,第三代钙离子拮抗剂阿罗地平和第三代 B 受体拮抗剂如卡维地洛能降低DCM 患者心血管病病死率,增加存活率。 2.扩心病的发病机制?(1)急性病毒性心肌炎后病毒持续性感染 (2)自身免疫导致心肌进行性损害
)。 张和收缩功能受损为体征。 3.特异性心肌病是指与特异性心脏病或系统性疾病有关 的心肌疾病,常见的有( )等。 1.扩心病的的治疗原则: 2.扩心病的发病机制? 姓名: 分数: 一、填空题: (每题 5 分,共 16*5=80 分) 2015 年三基三严培训:心肌病的分类 试题 1.心肌病的分类:( )( 2.扩张型心肌病( DCM ):是以( )或( )( 4.扩张性心肌病占心衰人数的( ),死亡率高, 10 年生存率(10-30%)。出现心衰症状后, 5 年生存率 ( 5.扩心病常见的症状是 、问答题(共 10*2=20 ( 分) )(
张辉中国扩张型心肌病诊断和治疗指南·365医学网 ??2018年4月21日,我国首部《中国扩张型心肌病诊断和治疗指南》在第九届国际心血管病靶向治疗论坛暨第八届中部心脏病学会议上正式发布。协作组在2007年心肌病诊断与治疗建议基础上,引用国内外临床研究资料,借鉴国外指南和科学声明的优点,进行指南制定。 ??廖玉华教授在大会中指出,近年来,社会公众对于高血压、冠心病等心血管病的防治工作已开始重视,但对于心肌病及其可造成的健康危害却知之甚少,我们应该像重视高血压一样重视扩心病防治。 中国扩张型心肌病诊疗指南的总体原则 ??·协作组在2007年《心肌病诊断与治疗建议》中DCM部分的基础上,引用国内外临床研究资料,借鉴国外指南和科学声明的优点进行本指南的制定。 ??·指南提出的推荐意见是在系统评估基础上由协作组专家讨论形成,当意见出现分歧时,在充分考虑不同意见的基础上接受多数专家的共识。 ??本指南的推荐类别和级别定义借鉴欧美心衰指南 下面我们将指南从以下几点具体展开: ??一、扩张型心肌病的定义与病因分类 ??二、扩张型心肌病的生物标记物
??三、扩张型心肌病的影像学检查 ??四、扩张型心肌病的诊断标准 ??五、扩张型心肌病的治疗原则 ??六、扩张型心肌病的药物与非药物治疗 ??七、扩张型心肌病特殊类型的诊治要点 ??八、扩张型心肌病的心脏康复治疗 一、扩张型心肌病的定义和病因分类 ?? 1.定义:DCM是一种异质性心肌病,以心室扩大和心肌收缩功能降低为特征,发病时除外高血压、心脏瓣膜病、先天性心脏病或缺血性心脏病等。 流行病学: ??发病率:年5-8/10万 ??好发人群:黑人和男子多发,是白人和女子的3倍以上??发病年龄:20-60岁 临床表现: ??症状:左心功能不全表现 ??呼吸困难 ??咳嗽,咳痰,咯血 ??乏力,疲劳,头昏,心慌 ??少尿和肾功不全表现 ??右心功能不全表现
《超声心动图诊断心肌病临床应用指南》(2020)要点 心肌病是一类病因和临床表现均比较复杂的疾病,近年来发病率呈上升趋势。各种影像技术如心脏磁共振(CMR)、心脏CT和超声心动图等在心肌病的诊断和分类中发挥重要作用。超声心动图技术操作简单,可直观显示心脏形态结构改变、室壁运动、心脏血流和功能情况,在心肌病的诊断、危险分层及预后判断中具有重要价值,成为心肌病检查及随访的首选检查方法。近年来超声声学增强剂的应用、实时三维超声心动图(RT-3DE)和斑点追踪成像(STI)等技术的迅猛发展,为心肌病的诊断提供了更多重要信息,结合基因检测和临床其他检查极大提高了心肌病患者的病因诊断水平。 扩张型心肌病 一、扩张型心肌病概述 扩张型心肌病(DCM)是一种原发的由心肌功能障碍引起的疾病,表现为左心室或双心室扩大、收缩功能障碍、无其他负荷异常(高血压、瓣膜病等)或冠状动脉病变。 二、诊断标准 超声心动图检查是DCM诊断的首选影像学技术,X线胸片、CMR、心脏
CT同样有助于诊断。在对DCM诊断时,需要排除引起心肌损害的其他疾病。临床常用诊断标准:左心室舒张期末内径(LVEDd)>5.0cm(女性)和>5.5cm (男性)(或相对准确方法:经体表面积校正的LVEDd>2.7cm/m2,或LVEDd大于年龄和体表面积预测值的117%,即预测值的2倍标准差+5%);左心室射血分数(LVEF)<45%和(或)左心室短轴缩短速率(LVFS)<25%;除外高血压、瓣膜病、先天性心脏病和缺血性心脏病等。 三、超声心动图检查 1. 心脏结构改变 2. 心脏收缩功能明显减低 3. 瓣膜反流 4. 左心室充盈压升高 5. 心腔声学造影 6. 负荷超声心动图检查
2015年三基三严培训:心肌病的分类试题 姓名:分数: 一.填空题:(每题5分,共16*5=80分) 1.心肌病的分类:(扩张型心肌病)(肥厚型心肌病)(限制型心肌病)(致 心律失常性右室发育不全心肌病)(未分化的心肌病)(特异性心肌病) 2.扩张型心肌病(DCM):是以(左心室)或(双心室)扩张和收缩功能受损为 体征。 3.特异性心肌病是指与特异性心脏病或系统性疾病有关的心肌疾病,常见的有(缺血 性心肌病)(瓣膜性心肌病)(高血压性心肌病)等。 4.扩张性心肌病占心衰人数的( 10-15%),死亡率高,10年生存率(10-30%)。出现 心衰症状后,5年生存率(40%) 5.扩心病常见的症状是( 室性心律失常 )(猝死)。 二、问答题(共10*2=20分) 1.扩心病的的治疗原则: 强心、利尿、ACEI等药物治疗外,第三代钙离子拮抗剂阿罗地平和第三代B受体拮抗剂如卡维地洛能降低DCM患者心血管病病死率,增加存活率。 2.扩心病的发病机制? (1)急性病毒性心肌炎后病毒持续性感染
(2)自身免疫导致心肌进行性损害 2015年三基三严培训:心肌病的分类试题 姓名:分数: 一、填空题:(每题5分,共16*5=80分) 1.心肌病的分类:()()()()() ()。 2.扩张型心肌病(DCM):是以()或()扩张和收缩功能受损为体征。 3.特异性心肌病是指与特异性心脏病或系统性疾病有关的心肌疾病,常见的有 ()()()等。 4.扩张性心肌病占心衰人数的(),死亡率高,10年生存率(10-30%)。出现心 衰症状后,5年生存率() 5.扩心病常见的症状是( )()。 二、问答题(共10*2=20分) 1.扩心病的的治疗原则: 2.扩心病的发病机制?
第二节病毒性心肌炎 【ICD-10编码】I40.001 【定义】 病毒性心肌炎是指病毒侵犯心脏,以心肌炎性病变为主要表现的疾病,有时病变可累及心包或心内膜。 【病因】 引起儿童心肌炎常见的病毒有柯萨奇病毒(B组和A组)、埃可(ECHO)病毒、脊髓灰质炎病毒、腺病毒、流感和副流感病毒、麻疹病毒、流行性腮腺炎病毒、传染性肝炎病毒等,新生儿期柯萨奇病毒B组感染可导致群体流行,死亡率高。【诊断要点】 1.症状和体征 (1)症状:表现轻重不一,取决于年龄与感染的急性或慢性过程,预后大多良好。大多数患儿有发热、咽痛、咳嗽等上呼吸道病毒感染或腹痛、腹泻等消化道病毒感染等前驱症状。心脏受累轻者可无症状或有胸闷、胸痛、心悸、乏力,活动受限等症状,少数重症可发生心力衰竭并严重心律失常、心源性休克,猝死。新生儿患病病情进展快,常见高热、反应低下、呼吸困难、发绀,常有神经、肝脏和肺的并发症。 (2)体征:心脏有轻度扩大,伴心动过速,偶有心动过缓、心律不齐、心音低钝及奔马律。有心包炎者可闻及心包摩擦音。重症病例反复心衰者,心脏明显扩大,肺部出现湿啰音及肝、脾肿大,呼吸急促和紫绀,重症患者可突然发生心源性休克,脉搏细弱,血压下降。 2.辅助检查 (1)X线检查:心影大小正常或增大,严重者有肺淤血或水肿,少数可伴有心包积液。 (2)心电图:可见严重心律失常,包括各种期前收缩,室上性和室性心动过速,房颤和室颤,II度和III度房室传导阻滞。心肌明显受累时可见T波降低、ST 段改变等。心电图缺乏特异性,应动态观察。 (3)超声心动图:轻者无改变。重者可有心房、心室扩大,以左心室扩大为主,或有心包积液、胸腔积液、心力衰竭者心脏收缩功能减退。
肥厚型心肌病诊断及治疗指南 一、肥厚型心肌病的定义和流行病学 1958年Teare首先对“肥厚型心肌病”进行了详细描述,随后概念不断演变发展,该病基本特征是心肌肥厚及猝死发生率高。目前认为,HCM是一种以心肌肥厚为特征的心肌疾病,主要表现为左心室壁增厚,通常指二维超声心动图测量的室间隔或左心室壁厚度≥15mm,或者有明确家族史者厚度≥13mm,通常不伴有左心室腔的扩大,需排除负荷增加如高血压、主动脉瓣狭窄和先天性主动脉瓣下隔膜等引起的左心室壁增厚。 一些来源于特定人群的患病率调查发现HCM并不少见。中国HCM患病率为80/10万,粗略估算中国成人HCM患者超过100万。HCM是青少年和运动员猝死的主要原因之一。心脏性猝死(SCD)常见于10~35岁的年轻患者,心衰死亡多发生于中年患者,HCM相关的心房颤动(房颤)导致的卒中则以老年患者多见。在三级医疗中心就诊的HCM患者年死亡率为2%~4%,SCD是最常见的死因之一。 二、肥厚型心肌病的分型 根据超声心动图检查时测定的左心室流出道与主动脉峰值压力阶差(LVOTG),可将HCM患者分为梗阻性、非梗阻性及隐匿梗阻性3种类型。安静时LVOTG≥30mmHg为梗阻性;安静时LVOTG正常,负荷运动时LVOTG≥30mmHg为隐匿梗阻性;安静或负荷时LVOTG 均<30mmHg为非梗阻性。 另外,约3%的患者表现为左心室中部梗阻性HCM,可能无左心室流出道梗阻,也无收缩期二尖瓣前向运动(SAM)征象。有研究认为这类患者的临床表现及预后与梗阻性HCM 相同,甚至更差。 梗阻性、隐匿梗阻性和非梗阻性HCM患者比例约各占1/3。这种分型有利于指导治疗方案选择,是目前临床最常用的分型方法。 此外根据肥厚部位,也可分为心尖肥厚、右心室肥厚和孤立性乳头肌肥厚的HCM。2013年世界心脏基金会对心肌病采用了新的综合分型系统,称为MOGE(S)分型。该分型保留了对心脏形态功能的识别,同时强调了疾病的遗传基础,但应用尚不成熟,仅供参考。 三、肥厚型心肌病的诊断 1.症状 HCM临床症状变异性大,有些患者可长期无症状,而有些患者首发症状就是猝死。儿童或青年时期确诊的HCM患者症状更多、预后更差。症状与左心室流出道梗阻、心功能受损、快速或缓慢型心律失常等有关,主要症状如下。
49 中国循环杂志 2019年11月 第34卷 Chinese Circulation Journal,November,2019,Vol. 34 Supplment AHA 儿童心肌病的分类和诊断科学声明解读 傅立军,张浩 作者单位:200127 上海市,国家儿童医学中心 上海交通大学医学院附属上海儿童医学中心 心脏中心通信作者:张浩 Email: drzhanghao@https://www.doczj.com/doc/a59418429.html, 中图分类号:R54 文献标识码:C 文章编号:1000-3614(2019)增刊-0049-05 DOI:10.3969/j.issn.1000-3614.2019. 增刊.011关键词 儿童;心肌病;分子遗传性疾病;分类 心肌病是以心肌病变为主要表现的一组异质性疾病,在儿童时期并不多见,但预后极差,是引起儿童心功能不全和心原性猝死的常见原因之一。据国外研究资料,近40%有症状的心肌病患儿在诊断后2年内接受心脏移植或者死亡。过去10年中儿童心肌病的诊治虽然取得了一些进展,但其预后尚未得到根本性的改善,罹患心肌病的儿童接受心脏移植的比例仍居高不下,心肌病依然是1岁以上儿童心脏移植的首要原因。目前在儿童心肌病的诊治中, 还存在着很多亟待解决的问题:心肌病的病因众多,病因诊断对于心肌病预后的判断以及治疗方案的选择具有重要的指导意义,但在临床上仅有少数心肌病患儿能明确其具体病因,对于大多数患儿仍缺乏对其病因的精准诊断;已发表的儿童心肌病相关的临床研究非常少见,相关的专题学术会议也少有开展,儿童心肌病的循证医学依据非常有限;儿童心肌病与成人心肌病虽有很多类似之处,但也有自身特点,目前儿童心肌病的分类和诊断策略主要借鉴于成人经验,迄今为止仍没有儿童心肌病分类和诊断策略的指导性文件。近年来,随着心肌病相关的基础、转化和临床 研究的进展,尤其是基因组学技术所推动的精准医疗的发展,越来越多的不明原因的心肌病被揭开了神秘的面纱,从而对儿童心肌病的病因有了更为深入的认识,并为儿童心肌病的诊断策略带来了新的变化。为此,美国心脏协会(AHA)组织儿童心肌病领域的相关专家,对目前儿童心肌病病因方面的最新认识和最优诊断策略进行了总结,并于2019年7月在《循环》杂志上在线发表了《儿童心肌病的分类和诊断:AHA 科学声明》(以下简称《科学声明》),主要内容涉及儿童心肌病的发病率、分类、形态学评估、分子遗传检测以及各种类型的儿童心肌病的病因和诊断方法,是首个关于儿童心肌病分类和诊断的共识声明,为儿童心肌病的临床诊断提供了重要指导和参考,并将有助于凝练未来儿童心肌病的优先研究方向,从而使该症患儿能得到早期精准诊断并进一步改善其临床预后。以下对其相关内容进行逐一解读。1 儿童原发性心肌病的发病率目前尚无儿童心肌病确切的发病率,相关的流行病学资料主要来自于西方国家。据美国、芬兰和澳大利亚的基于人口的研究资料,儿童原发性心肌病的年发病率约为十万分之一,但在年龄、性别和种族之间存在一定的差异,心肌病在婴儿期高发,其发病率较年长儿高8倍。在儿童心肌病中,以扩张型心肌病(DCM)最为多见(约占50%),肥厚型心肌病(HCM)次之(约占35%~50%),限制型心肌病(RCM)相对少见(不足5%),左心室心肌致密化不全(LVNC)约占5%。 2 儿童心肌病的分类心肌病的定义和分类是一个长期争议的问题,目前成人心肌病存在多种分类方法,包括1995年世界卫生组织(WHO)发布的基于形态功能的分类方法,2006年AHA 推荐的主要基于遗传学的分类方法,2008年欧洲心脏学会(ESC)推荐的主要基于形态功能的分类方法,以及2013年世界心脏联盟(WHF)推荐的包含临床表现及遗传学特点的MOGE(S)分类方法。对于儿童患者,在新生儿期、婴儿期、儿
中华心血管病杂志!##6 年" 月第5& 卷第" 期% .MH? S .EDJH>K,SE?QEDF !##6, 3>K8 5& T>8 " ? & ? ?对策研究? 心肌病诊断与治疗建议 中华医学会心血管病学分会% 中华心血管病杂志编辑委员会% 中国心肌病诊断与治疗建议工作组 % % "$$& 年世界卫生组织( '())* 国际心脏病学会联合会 (+,-.)将心肌病定义为伴心功能不全的心肌疾病,分为原发性和继发性二类["]。原发性心肌病包括扩张型心肌病(/.0)、肥厚型心肌病 ((.0)、致心律失常性右室心肌病 (123.)、限制型心肌病(2.0 )和未定型心肌病。十年来,该定义和分类被临床和病理医生广泛接受和应用。"$$$ 年"" 月中华心血管病杂志发表心肌炎和心肌病会议讨论纪要 建议我国临床医师采用上述标准[! ]。 由于心脏超声等影像技术的进步,分子生物学、分子遗 传学理论和知识的应用,多中心、大规模临床“ 循证医学”证
据的获得,近年来科学家和临床学家们对心肌病的发病、命名、诊断、治疗及预后发表了许多新的见解。& 年内可查阅心肌病有关的文献总量超过"4 ### 篇,中、英文文献"5 !6$ 篇,其中中文!56 篇。/.0 和(.0 的患病率分别达到578 & 和!## * "# 万人群,心肌病呈发病上升、年死亡率降低的趋势[59&]。影像发现提供诊断和分类依据,基因诊断和基因筛选近年已成为心肌病研究的新领域。临床治疗有多种选择,包括药物、介入、外科手术和心脏移植等方法,心肌病已成为可知原因、能够诊断和治疗的常见病。心肌病基因和基因后修饰资料的累积和发现,心肌肌节 蛋白基因突变导致(.0 发病超过&#: ,也有报道达到 ;&: ,因此(.0 被定义和分类为遗传性心肌疾病[79$]。青少 年和运动员猝死与基因突变相关,已列入心肌病的范畴,美已形成“ 标准” 和“指南”性文件["#] '() * +,-. 心肌病标准已不能涵盖和反映心肌病临床现实的需要,北美、 欧洲已发布了多个相关指南、明,起草专家们详细地阐述了(.0 、/.0 和123. 多个方面的进展,评估了基因筛选、基因诊断的现状和重要性,置入性心脏除颤起搏器( +./)可防治恶性心律失常导致的猝死,药物难治性(.0 的化学消融和外科干预,心脏再同步化治疗
2014年ESC 肥厚型心肌病诊断和治疗指南 肥厚型心肌病(Hypertrophic cardiomyopathy,HCM)是指并非完全因心脏负荷异常引起的左心室室壁增厚。虽然HCM 是一种常见的心血管疾病,但却几乎没有相应的随机对照临床研究。 所以,本次指南中的大部分推荐都基于观察性队列研究和专家的统一意见,为医生提供所有年龄段患者临床诊断和治疗概览,此外,由于大部分患者都有遗传病因参与,本次指南同时还纳入了对家庭成员进行诊断的内容,并对生殖和避孕做出了特别建议。 该定义对儿童与成人均可适用,并不需要对病因或心肌病理进行先验推测。虽然这个定义会扩大指南的范围,并使一些推荐复杂化,但与每日临床实践紧密相关,能更好地改善诊断的准确性和治疗。 一、病因 高达60% 的青少年与成人HCM 患者的病因是心脏肌球蛋白基因突变引起的常染色体显性遗传。5-10% 的成人患者病因为其他遗传疾病,包括代谢和神经肌肉的遗传病、染色体异常和遗传综合征。还有一些患者的病因是类似遗传疾病的非遗传疾病,如老年淀粉样变性。 二、诊断标准 1. 成人 成人中HCM 定义为:任意成像手段(超声心动图、心脏核磁共振成像或计算机断层扫描)检测显示,并非完全因心脏负荷异常引起的左室心肌某节段或多个节段室壁厚度≥15 mm。 遗传或非遗传疾病可能表现出来的室壁增厚程度稍弱(13-14 mm),对于这部分患者,需要评估其他特征以诊断是否为HCM,评估内容包括家族病史、非心脏性症状和迹象、心电图异常、实验室检查和多模式心脏成像。 2. 儿童 与成人中一样,诊断HCM 需要保证LV 室壁厚度≥ 预测平均值+ 2 SD(即Z 值>2,Z 值定义为所测数值偏离平均值的SD 数量)。 3. 亲属 对于HCM 患者的一级亲属,若心脏成像(超声心动图、心脏核磁共振或CT)检测发现无其他已知原因的LV 室壁某节段或多个节段厚度≥13 mm,即可确诊HCM。 在遗传性HCM 家族中,未出现形态学异常的突变携带者可能会出现心电图异常,这种异常的特异性较差,但在有遗传性HCM 的家族成员身上,可视为HCM 疾病的早期或温和表现,其他多种症状也可以提高对这部分人群诊断的准确性。 总而言之,对于遗传性HCM 的家族成员,任何异常(如心肌多普勒成像和应变成像异常、不完全二尖瓣收缩期前移或延长和乳头肌异常)尤其是心电图异常都会大大增加该成员诊断出HCM 的可能性。
ESC2014 肥厚型心肌病诊断和治疗指南 肥厚型心肌病(Hypertrophic cardiomyopathy,HCM)是指并非完全因心脏负荷异常引起 的左心室室壁增厚。虽然 HCM 是一种常见的心血管疾病,但却几乎没有相应的随机对照 临床研究。 所以,本次指南中的大部分推荐都基于观察性队列研究和专家的统一意见,为医生提供所 有年龄段患者临床诊断和治疗概览,此外,由于大部分患者都有遗传病因参与,本次指南 同时还纳入了对家庭成员进行诊断的内容,并对生殖和避孕做出了特别建议。 该定义对儿童与成人均可适用,并不需要对病因或心肌病理进行先验推测。虽然这个定义 会扩大指南的范围,并使一些推荐复杂化,但与每日临床实践紧密相关,能更好地改善诊 断的准确性和治疗。 一、病因 高达 60% 的青少年与成人 HCM 患者的病因是心脏肌球蛋白基因突变引起的常染色体显性 遗传。5-10% 的成人患者病因为其他遗传疾病,包括代谢和神经肌肉的遗传病、染色体异 常和遗传综合征。还有一些患者的病因是类似遗传疾病的非遗传疾病,如老年淀粉样变性。 二、诊断标准 1. 成人 成人中HCM 定义为:任意成像手段(超声心动图、心脏核磁共振成像或计算机断层扫描)检测显示,并非完全因心脏负荷异常引起的左室心肌某节段或多个节段室壁厚度≥ 15 mm。 遗传或非遗传疾病可能表现出来的室壁增厚程度稍弱(13-14 mm),对于这部分患者,需要评估其他特征以诊断是否为 HCM,评估内容包括家族病史、非心脏性症状和迹象、心 电图异常、实验室检查和多模式心脏成像。 2. 儿童 与成人中一样,诊断 HCM 需要保证 LV 室壁厚度≥ 预测平均值+ 2 SD(即 Z 值>2,Z 值 定义为所测数值偏离平均值的 SD 数量)。 3. 亲属 对于 HCM 患者的一级亲属,若心脏成像(超声心动图、心脏核磁共振或 CT)检测发现无其他已知原因的 LV 室壁某节段或多个节段厚度≥ 13 mm,即可确诊 HCM。 在遗传性 HCM 家族中,未出现形态学异常的突变携带者可能会出现心电图异常,这种异 常的特异性较差,但在有遗传性 HCM 的家族成员身上,可视为 HCM 疾病的早期或温和 表现,其他多种症状也可以提高对这部分人群诊断的准确性。 总而言之,对于遗传性HCM 的家族成员,任何异常(如心肌多普勒成像和应变成像异常、不完全二尖瓣收缩期前移或延长和乳头肌异常)尤其是心电图异常都会大大增加该成员诊 断出 HCM 的可能性。
此文档下载后即可编辑 第二节病毒性心肌炎 【ICD-10编码】I40.001 【定义】 病毒性心肌炎是指病毒侵犯心脏,以心肌炎性病变为主要表现的疾病,有时病变可累及心包或心内膜。 【病因】 引起儿童心肌炎常见的病毒有柯萨奇病毒(B组和A组)、埃可(ECHO)病毒、脊髓灰质炎病毒、腺病毒、流感和副流感病毒、麻疹病毒、流行性腮腺炎病毒、传染性肝炎病毒等,新生儿期柯萨奇病毒B组感染可导致群体流行,死亡率高。 【诊断要点】 1.症状和体征 (1)症状:表现轻重不一,取决于年龄与感染的急性或慢性过程,预后大多良好。大多数患儿有发热、咽痛、咳嗽等上呼吸道病毒感染或腹痛、腹泻等消化道病毒感染等前驱症状。心脏受累轻者可无症状或有胸闷、胸痛、心悸、乏力,活动受限等症状,少数重症可发生心力衰竭并严重心律失常、心源性休克,猝死。新生儿患病病情进展快,常见高热、反应低下、呼吸困难、发绀,常有神经、肝脏和肺的并发症。 (2)体征:心脏有轻度扩大,伴心动过速,偶有心动过缓、心律不齐、心音低钝及奔马律。有心包炎者可闻及心包摩擦音。重症病例反
复心衰者,心脏明显扩大,肺部出现湿啰音及肝、脾肿大,呼吸急促和紫绀,重症患者可突然发生心源性休克,脉搏细弱,血压下降。 2.辅助检查 (1)X线检查:心影大小正常或增大,严重者有肺淤血或水肿,少数可伴有心包积液。 (2)心电图:可见严重心律失常,包括各种期前收缩,室上性和室性心动过速,房颤和室颤,II度和III度房室传导阻滞。心肌明显受累时可见T波降低、ST段改变等。心电图缺乏特异性,应动态观察。(3)超声心动图:轻者无改变。重者可有心房、心室扩大,以左心室扩大为主,或有心包积液、胸腔积液、心力衰竭者心脏收缩功能减退。 (4)实验室检查:白细胞数增高,血沉增快,谷草转氨酶、乳酸脱氢酶、磷酸激酶及其同工酶活性增高,肌钙蛋白阳性。 (5)病原学检查:以咽拭子,粪便、尿液、血液、心包液进行病毒分离,或者在恢复期做血清补体结合试验、中和试验,可有特异性病毒抗体明显升高。 【诊断标准】 根据1999年中华医学会儿科学分会心血管学组修订后的小儿病毒性心肌炎诊断标准: 1、临床诊断依据: (1)心功能不全、心源性休克或心脑综合征。 (2)心脏扩大(X线、超声心动图检查具有表现之一)。
特发性心肌病诊疗指南 概述 心肌病是一组异质性心肌疾病。由各种不同原因(常为遗传原因)引起,伴有心肌机械和(或)心电活动障碍,常表现为不适当心室肥厚或扩张,可导致心功能不全或心血管死亡。原发性心肌病是指病变仅局限在心肌,根据发病机制,又分为遗传性、遗传和非遗传混合性及获得性3种。此处特发性心肌病(idiopathic cardiomyopathy)主要指以遗传性为主(包括混合性)的心肌病,包括特发性或家族性扩张型心肌病、致心律失常型右室发育不良/心肌病、特发性或者家族性限制型心肌病、左室致密化不全以及遗传性转甲状腺素蛋白相关心肌淀粉样变。扩张型心肌病(dilated cardiomyopathy,DCM)是指以左室或双心腔扩大和收缩功能障碍等为特征的一种疾病,其中约50%无法明确病因,将其定义为特发性DCM(idiopathic dilated cardiomyopathy),对这些患者进行3~4代详细的家族史询问并对一级亲属进行临床筛查,发现其中20%~35%患者具有基因突变和家族遗传背景,被称作家族性扩张型心肌病(familial dilated cardiomyopathy,FDCM)。致心律失常性右室发育不良/心肌病(arrhythmogenic right ventricular dysplasia/ cardiomyopathy,ARVD/C)是一种以心律失常、心力衰竭及心源性猝死为主要表现的非炎性、非冠状动脉心肌疾病,病理特点为右室心肌细胞被脂肪或纤维脂肪组织进行性取代,致使右室弥漫性扩张、收缩运动减弱。限制型心肌病(restrictive cardiomyopathy,RCM)是最为少见的心肌病,通常由于室壁僵硬,导致严重的舒张期功能障碍和充盈受限,临床表现为以右心为主的全心衰。绝大多数患者心室无扩张、室壁厚度正常且左室收缩功能正常。RCM可以为特发、家族性或者系统性疾病所致。此处指特发和家族性RCM。左室致密化不全(left ventricular non-compaction,LVNC)是一种以左室心肌小梁突出和小梁间隐窝深陷为特征,导致收缩和舒张功能障碍、传导异常和血栓栓塞事件的心肌疾病。孤立性LVNC是指不合并其他心脏或非心脏先天性异常的情况下发生的LVNC。遗传性转甲状腺素蛋白相关淀粉样变(hereditary transthyretin amyloidosis,hATTR)是常染色体显性遗传,由转甲状腺素蛋白基因突变产生异常TTR蛋白沉积在多个组织器官,导致淀粉样变,以进行性神经病变和心肌病为主要特征。. 病因和流行病学 FDCM大多为常染色体显性遗传,但各种遗传方式都有(常染色体隐性遗传、X 连锁遗传和线粒体遗传)。目前已经发现30多个基因突变,多为肌节蛋白基因突变。ARVC家族性发病占30%~50%,已经证实主要为编码桥粒的基因突变所致。家族性RCM常以常染色体显性遗传为特征,主要累及肌节蛋白基因,少数可能与常染色体隐性遗传(如HFE基因突变或糖原贮积病引起的血色素沉着症)有关,或与X连锁遗传有关。孤立性LVNC发病机制尚不清楚,一般认为是胚胎发育过程中胎儿心肌原基的疏松网状组织致密化过程停滞所致。直到现在,孤立性LVNC到底是一种独立的疾病还是其他心肌病的特殊表现还有争议,12%~50%的LVNC患者有家族史,其遗传方式大多为常染色体显性遗传,也可为X连锁遗传或常染色体隐性遗传。遗传性ATTR是TTR基因突变所致,目前已知突变类型超过120种。
肥厚型心肌病(Hypertrophic cardiomyopathy,HCM)是指并非完全因心脏负荷异常引起的左心室室壁增厚。虽然HCM 是一种常见的心血管疾病,但却几乎没有相应的随机对照临床研究。 所以,本次指南中的大部分推荐都基于观察性队列研究和专家的统一意见,为医生提供所有年龄段患者临床诊断和治疗概览,此外,由于大部分患者都有遗传病因参与,本次指南同时还纳入了对家庭成员进行诊断的内容,并对生殖和避孕做出了特别建议。 该定义对儿童与成人均可适用,并不需要对病因或心肌病理进行先验推测。虽然这个定义会扩大指南的范围,并使一些推荐复杂化,但与每日临床实践紧密相关,能更好地改善诊断的准确性和治疗。 一、病因 高达60% 的青少年与成人HCM 患者的病因是心脏肌球蛋白基因突变引起的常染色 体显性遗传。5-10% 的成人患者病因为其他遗传疾病,包括代谢和神经肌肉的遗传病、染色体异常和遗传综合征。还有一些患者的病因是类似遗传疾病的非遗传疾病,如老年淀粉样变性。 二、诊断标准 1. 成人 成人中HCM 定义为:任意成像手段(超声心动图、心脏核磁共振成像或计算机断层扫描)检测显示,并非完全因心脏负荷异常引起的左室心肌某节段或多个节段室壁厚度≥15 mm。 遗传或非遗传疾病可能表现出来的室壁增厚程度稍弱(13-14 mm),对于这部分患者,需要评估其他特征以诊断是否为HCM,评估内容包括家族病史、非心脏性症状和迹象、心电图异常、实验室检查和多模式心脏成像。 2. 儿童 与成人中一样,诊断HCM 需要保证LV 室壁厚度≥ 预测平均值+ 2 SD(即Z 值>2,Z 值定义为所测数值偏离平均值的SD 数量)。 3. 亲属 对于HCM 患者的一级亲属,若心脏成像(超声心动图、心脏核磁共振或CT)检测发现无其他已知原因的LV 室壁某节段或多个节段厚度≥13 mm,即可确诊HCM。 在遗传性HCM 家族中,未出现形态学异常的突变携带者可能会出现心电图异常,这种异常的特异性较差,但在有遗传性HCM 的家族成员身上,可视为HCM 疾病的早期或温和表现,其他多种症状也可以提高对这部分人群诊断的准确性。 总而言之,对于遗传性HCM 的家族成员,任何异常(如心肌多普勒成像和应变成像异常、不完全二尖瓣收缩期前移或延长和乳头肌异常)尤其是心电图异常都会大大增加该成员诊断出HCM 的可能性。 三、诊断 1. 静息和动态心电图检查建议 建议分级水平参考文献
心脏病、先心病的资料、预防、分类、救助、遗传。 关于心脏病的分类:(转自 39 心血管科)核心提示:心脏病的种类有哪些? 专题表示, 心脏病主要有以下八个种类 (补充) , 且不同心脏病之间症状与起因也各不相同。 心脏病的种类有哪些? 心脏病是心脏疾病的总称。 心脏病的种类有很多, 下面我们来具体讲解一下心脏病究竟有哪 些种类,各类心脏病又有哪些不同呢? 专家谈到,心脏病主要有以下八个种类,且不同心脏病之间症状与起因也各不相同。
心脏病的种类一 风湿性心瓣膜病: 也称慢性风湿性心脏病, 是指急性风湿性心脏炎后所遗留下来的以心脏各 瓣膜病变为主的一种心脏病。 心脏病的种类二 先天性心脏:一些染色体异常的疾病常伴有先天性心脏病,家族遗传史。 心脏病的种类三 冠心病:抽烟及糖尿病、高血压等导致血管硬化狭窄,使血流受阻,易使心肌缺氧而受损。 劳累或精神紧张时出现胸骨后或心前区疼痛或紧缩样疼痛;体力活动出现胸闷、 心悸、 气短; 出现与运动有关的头痛、 牙痛、 腿痛;饱餐、 寒冷或看惊险片时胸痛、 心悸;平卧时突然胸痛、 心悸、呼吸困难;性生活或排便困难时出现心慌、胸闷等不适。 心脏病的种类四 高血压心脏病:动脉性高血压会导致左心室肥大;肺高压症会导致右心室肥大。由于血压长 期升高,心脏的左心室泵血阻力上升,左心室长期处于超负荷状态,因代偿而逐渐肥厚、扩 张,心肌耗氧增加,心肌重量增加,但无相应的供血增加。 同时, 高血压损害冠状动脉血管, 发生粥样硬化,使供应心肌的血液减少。两者联合作用,会导致心律紊乱、心绞痛、心肌梗 死、心力衰竭等。
心脏病的种类五 风湿性心脏病: 风湿性心瓣膜病也称慢性风湿性心脏病, 是指急性风湿性心脏炎后所遗留下 来的以心脏各瓣膜病变为主的一种心脏病。心悸气短,活动后喘促、疲劳,乏力,咯血等左 心功能不全。重者出现头昏,心绞痛,心律失常,甚至晕厥,猝死。晚期出现呼吸困难、咳 嗽、咯血等左心功能不全症状,体征为主动瓣区听到响亮粗糙的吹风样收缩期杂音,向颈部 传导,并伴有收缩期震颤等。 心脏病的种类六 肺源性心脏病:简称肺心病,是由于各种胸肺及支气管病变而继发的肺动脉高压,最后导致 以右室肥大为特点的心脏病。大多数肺心病是从慢性支气管炎、阻塞性肺气肿发展而来,少 部分与支气管哮喘、肺结核、支气管扩张有关。肺源性心脏病常年存在,多于冬春季节并发 呼吸道感染而导致呼吸衰竭和心力衰竭,病死率较高。 心脏病的种类七
心肌、心脏肿瘤及血管病变:新陈代谢或荷尔蒙异常的心肌变化等,有时酗酒,药物亦导致 心肌变化。心脏肿瘤大多为良性肿瘤,以黏液瘤为最常见,原发性心脏恶性肿瘤很少见。血 管病变包括高血压引起之动脉瘤, 以及其他免疫机能异常引起之血管病变等。 多见于老年性 心脏病。 进行治疗。 心脏疾病有很多,由于发生的部位和程度不同而分为各式各样的类型。以上为 心脏病的种类。 我们了解到不同心脏病之间症状与起因也各不相同。 患者应根据自己的类型
心脏病的种类八 扩张性心脏病:扩张型心肌病的大部分患者不能找到明确的发病原因,属于特发性,另一部 分患者的发病与病毒感染、自身免疫反应、饮酒、中毒、家族遗传因素等有关。
(1)
?
? ? ?
打赏
回复 1楼 2014-01-21 16:49
52.特发性心肌病 概述 心肌病是一组异质性心肌疾病。由各种不同原因(常为遗传原因)引起,伴有心肌机械和(或)心电活动障碍,常表现为不适当心室肥厚或扩张,可导致心功能不全或心血管死亡。原发性心肌病是指病变仅局限在心肌,根据发病机制,又分为遗传性、遗传和非遗传混合性及获得性3 种。此处特发性心肌病(idiopathic cardiomyopathy)主要指以遗传性为主(包括混合性)的心肌病,包括特发性或家族性扩张型心肌病、致心律失常型右室发育不良/心肌病、特发性或者家族性限制型心肌病、左室致密化不全以及遗传性转甲状腺素蛋白相关心肌淀粉样变。 扩张型心肌病(dilated cardiomyopathy,DCM)是指以左室或双心腔扩大和收缩功能障碍等为特征的一种疾病,其中约50%无法明确病因,将其定义为特发性DCM(idiopathic dilated cardiomyopathy),对这些患者进行3~4 代详细的家族史询问并对一级亲属进行临床筛查,发现其中20%~35%患者具有基因突变和家族遗传背景,被称作家族性扩张型心肌病(familial dilated cardiomyopathy,FDCM)。致心律失常性右室发育不良/心肌病(arrhythmogenic right ventricular dysplasia/ cardiomyopathy,ARVD/C)是一种以心律失常、心力衰竭及心源性猝死为主要表现的非炎性、非冠状动脉心肌疾病,病理特点为右室心肌细胞被脂肪或纤维脂肪组织进行性取代,致使右室弥漫性扩张、收缩运动减弱。限制型心肌病(restrictive cardiomyopathy,RCM)是最为少见的心肌病,通常由于室壁僵硬,导致严重的舒张期功能障碍和充盈受限,临床表现为以右心为主的全心衰。绝大多数患者心室无扩张、室壁厚度正常且左室收缩功能正常。RCM 可以为特发、家族性或者系统性疾病所致。此处指特发和家族性RCM。左室致密化不全(left ventricular non-compaction,LVNC)是一种以左室心肌小梁突出和小梁间隐窝深陷为特征,导致收缩和舒张功能障碍、传导异常和血栓栓塞事件的心肌疾病。孤立性LVNC 是指不合并其他心脏或非心脏先天性异常的情况下发生的LVNC。遗传性转甲状腺素蛋白相关淀粉样变(hereditary transthyretin amyloidosis,hATTR)是常染色体显性遗传,由转甲状腺素蛋白基因突变产生异常TTR 蛋白沉积在多个组织器官,导致淀粉样变,以进行性神经病变和心肌病为主要特征。
2018ESC图解第四版心肌梗死通用定义:心肌损伤+缺血证据才诊断心梗 与第三版相比,第四版心肌梗死通用定义内容明显更为充实,毕竟页数从原先的17页几乎增加了一倍达到了33页。但是从定义本身来讲,新一版的心肌梗死通用定义并没有有太大的改动,可以算是“没有显著性差异”。反过来讲,这也是一件好事,定义的稳定也反映了目前对心梗的认识是相对全面和准确的。 新一版心肌梗死通用定义依然延续了上一版的“1+1”诊断策略,仍然将肌钙蛋白(cTn)的动态变化,且至少有一次超过99%参考上限作为急性心梗(1型和2型)诊断中那不可缺少的“1”,并将这种cTn的变化明确作为一个新的概念提出,即急性心肌损伤(acute myocardial injury)。 ESC2018上,第四版心肌梗死通用定义发布。《2018年心肌梗死通用定义》较2014年版共识提出了5个新概念,更新了14个概念,增加了6个临床相关内容。 区分心肌梗死与心肌损伤:是否存在缺血是关键 心肌损伤:当心肌肌钙蛋白(cTn)升高,超过了正常值,就是心肌损伤。 慢性和急性心肌损伤:如果肌钙蛋白值存在升高和或下降过程,是急性心肌损伤。如果肌钙蛋白持续升高,就是慢性心肌损伤。 图1 急性心肌损伤(实线)和慢性心肌损伤(虚线) 在那些就诊较早的急性心梗患者,只要症状和心电图典型,并不一定需要等到cTn升高才能确诊急性心梗,才能按照心梗进行血运重建,所以在急诊有时并不需要死扣诊断标准。 其次,对于那些就诊较晚的心梗患者,初次cTn已经超过99%参考上限了,可是连续的2次cTn检查结果有可能刚好落在曲线的左侧和右侧相对称的点,此时两次cTn可以并没有动态改变,但是不能因此而排除心梗的诊断,此时需综合其他临床情况,适当延长检测