CHIP染色质免疫共沉淀实验方法简介
- 格式:ppt
- 大小:754.00 KB
- 文档页数:15

实验染色质免疫沉淀技术(ChIP)实验操作指南全心全意就为医生服务,一心一意只为造福医生。
真核生物的基因组DNA以染色质的形式存在。
因此,研究蛋白质与DNA在染色质环境下的相互作用是阐明真核生物基因表达机制的基本途径。
染色质免疫沉淀技术(chromatin immunoprecipitation assay, CHIP)是目前最常用研究体内DNA与蛋白质相互作用的方法。
它的基本原理是在活细胞状态下固定蛋白质-DNA复合物,并将其随机切断为一定长度范围内的染色质小片段,然后通过免疫学方法沉淀此复合体,特异性地富集目的蛋白结合的DNA片段,通过对目的片断的纯化与检测,从而获得蛋白质与DNA相互作用的信息。
CHIP不仅可以检测体内反式因子与DNA的动态作用,还可以用来研究组蛋白的各种共价修饰与基因表达的关系。
而且,CHIP与其他方法的结合,扩大了其应用范围:CHIP与基因芯片相结合建立的CHIP-on-chip方法已广泛用于特定反式因子靶基因的高通量筛选;CHIP与体内足迹法相结合,用于寻找反式因子的体内结合位点;RNA-CHIP用于研究RNA在基因表达调控中的作用。
由此可见,随着CHIP的进一步完善,它必将会在基因表达调控研究中发挥越来越重要的作用。
实验准备实验材料:细胞样品试剂、试剂盒:甲醛、甘氨酸、PBS、SDS Lysis Buffer、洗脱液、RNaseA、蛋白酶K、omega胶回收试剂盒仪器、耗材:离心管、超声仪、电泳仪、离心机ChIP的一般流程甲醛处理细胞→ 收集细胞,超声破碎→ 加入目的蛋白的抗体,与靶蛋白-DNA复合物相互结合→ 加入ProteinA,结合抗体-靶蛋白-DNA复合物,并沉淀→ 对沉淀下来的复合物进行清洗,除去一些非特异性结合→ 洗脱,得到富集的靶蛋白-DNA复合物→ 解交联,纯化富集的DNA-片断→ PCR分析。
请ChIp具体操作流程:第一天(一)、细胞的甲醛交联与超声破碎1、取出1平皿细胞(10cm平皿),加入243 μl 37%甲醛,使得甲醛的终浓度为1%(培养基共有9ml)。

染色质免疫沉淀分析——植物ChIP解决方案染色质免疫沉淀分析(ChIP) 是目前确定与特定蛋白结合的基因组区域或确定与特定基因组区域结合的蛋白质的比较好的一种方法。
so,我们今天来聊聊染色质免疫沉淀分析方法。
染色质免疫沉淀法(Chromatin immunoprecipitation,ChIP)是研究体内DNA与蛋白质相互作用的重要工具。
它可以灵敏地检测目标蛋白与特异DNA片段的结合情况,还可以用来研究组蛋白与基因表达的关系。
CHIP技术通过三大步骤实现:第一,甲醛固定后染色质分离和断片;第二,运用特异蛋白质抗体(CHIP级别),免疫共沉淀结合蛋白的染色质片段;第三,分析目标DNA。
这里我们就要说到CHIP技术工具之——植物染色质免疫沉淀试剂盒了。
它的原理是什么呢?不妨以P-2014植物染色质免疫沉淀试剂盒来举个栗子~~P-2014植物染色质免疫沉淀试剂盒旨在通过甲醛交联、物理或化学处理细胞核以及染色质,从而运用特异抗体结合蛋白进行免疫沉淀,然后萃取DNA,扩增分析,用以确定结合蛋白的目标DNA。
接下来再说说它的特征,P-2014植物染色质免疫沉淀试剂盒涵盖全套试剂,允许试验者有效地在体内研究蛋白-DNA相互关系。
整个过程可以在6小时内完成(哇哦,厉害了~)当然了,还有相当重要的一点:P-2014植物染色质免疫沉淀试剂盒适用于将特异性免疫沉淀与定性和定量PCR、MS-PCR、ChIP-Seq、ChIP-on-chip结合使用。
欧迈噶!P-2014植物染色质免疫沉淀试剂盒包括一个ChIP级二甲基组蛋白H3-K9抗体--阳性对照,以及一个正常小鼠IgG——阴性对照。
从染色质从样本中释放出来后,经剪切、断片,添加到包被了抗体的微孔中,蛋白质-DNA复合物经特异性抗体捕获,解交联后DNA被释放,通过离心柱,纯化并洗脱目的DNA。
洗脱下来的DNA可用于各种下游应用。
接着看看样本,起始材料可包括各种植物组织(花、叶、幼苗)。

染色体免疫共沉淀(Chip)实验报告步骤一:样品准备试剂和仪器:Biopulverizer(biospec)37% formaldehyde甘氨酸(Glycine)PBSprotease inhibitors步骤二:细胞交联1. 向客户提供的细胞沉淀中加入1ml 细胞培养基,混匀细胞后转移到15ml离心管中。
2. 向15ml离心管中加入270ul 37%甲醛溶液,使得甲醛的终浓度为1%,室温温育10min。
3. 向反应体系中各加入505ul 2.5M甘氨酸到终浓度为125mM,室温温育5min以终止交联反应。
4. 135x g,4°C离心10min,去上清,并用冰冷的10ml 1XPBS迅速漂洗两次。
5. 吸净PBS后,加入1ml PBS+protease inhibitors混合液,并转移到1.5ml离心管中。
800Xg,4°C离心5min,小心去掉上清。
步骤三:细胞裂解试剂:裂解缓冲液1: 50mM Hepes-KOH pH7.5; NaCl 140mM; EDTA 1mM; glycerol 10%;NP-40 0.5%;Tritonx -100 0.25%。
裂解缓冲液2: 10mM Tris-HCl pH8.0; NaCl 100mM; EDTA 1mM pH8.0; Na-Deoxycholate 0.1% Protease inhibitors。
步骤:1. 加入蛋白酶抑制剂(终浓度为1x) 到所有的裂解缓冲液中。
2. 用1ml的裂解缓冲液1重悬上述处理的样品,4°C旋转混合10min后,800g,4°C离心5min,弃上清。
3. 用300ul 裂解缓冲液2重悬样品,冰上放置30min。
步骤四:超声破碎DNA仪器:Bioruptor(Diagenode)步骤:(1)、将超声仪器Bioruptor 调到中档“Mid”(M)。
(2)、在超声池中注入一定量的冰水。

染色质免疫共沉淀原理染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP)是一种用于研究染色质中特定蛋白质与DNA相互作用的重要技术。
通过ChIP技术,可以确定染色质中特定蛋白质的结合位点,从而揭示基因调控网络和表观遗传调控机制。
本文将介绍染色质免疫共沉淀的原理及其在生物学研究中的应用。
ChIP技术的原理主要包括交联、裂解、免疫沉淀、洗涤和DNA纯化等步骤。
首先,将细胞进行交联,使得染色质中的蛋白质与DNA固定在一起。
随后,对细胞进行裂解,释放染色质蛋白质复合物。
接着,使用特异性抗体将目标蛋白质与其结合的DNA片段进行免疫沉淀。
然后,通过洗涤步骤去除非特异性结合的蛋白质和DNA,最终得到目标蛋白质结合的DNA片段。
最后,通过逆交联和DNA纯化,获得可以进一步分析的DNA样品。
ChIP技术在生物学研究中有着广泛的应用。
首先,ChIP技术可以用于确定染色质中特定蛋白质的结合位点,从而帮助研究人员理解基因的调控机制。
其次,ChIP技术还可以用于研究表观遗传调控,揭示组蛋白修饰与基因表达调控之间的关系。
此外,ChIP技术还可以用于筛选与某一特定蛋白质结合的DNA片段,从而帮助鉴定新的转录因子结合位点。
总之,ChIP技术在生物学研究中发挥着重要的作用,为研究人员提供了一种强大的工具来探究基因调控和表观遗传调控机制。
综上所述,染色质免疫共沉淀技术通过特异性抗体沉淀染色质中与特定蛋白质结合的DNA片段,为研究人员提供了一种重要的工具来研究基因调控和表观遗传调控。
随着生物学研究的不断深入,ChIP技术的应用也将变得更加广泛,为我们揭示生命的奥秘提供更多的可能性。
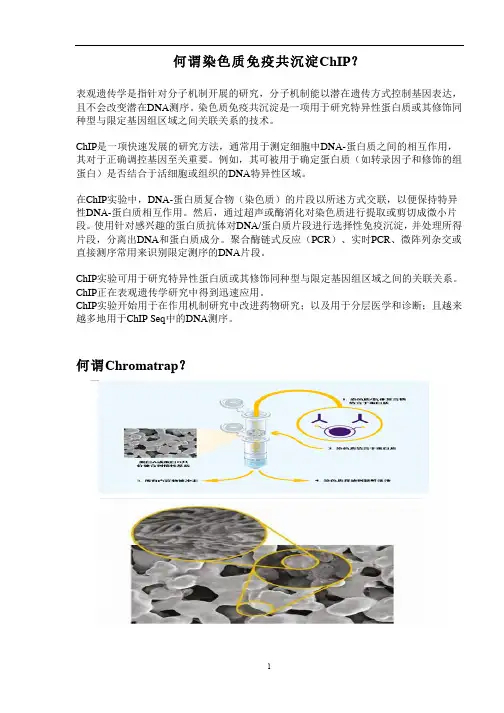
何谓染色质免疫共沉淀ChIP?表观遗传学是指针对分子机制开展的研究,分子机制能以潜在遗传方式控制基因表达,且不会改变潜在DNA测序。
染色质免疫共沉淀是一项用于研究特异性蛋白质或其修饰同种型与限定基因组区域之间关联关系的技术。
ChIP是一项快速发展的研究方法,通常用于测定细胞中DNA-蛋白质之间的相互作用,其对于正确调控基因至关重要。
例如,其可被用于确定蛋白质(如转录因子和修饰的组蛋白)是否结合于活细胞或组织的DNA特异性区域。
在ChIP实验中,DNA-蛋白质复合物(染色质)的片段以所述方式交联,以便保持特异性DNA-蛋白质相互作用。
然后,通过超声或酶消化对染色质进行提取或剪切成微小片段。
使用针对感兴趣的蛋白质抗体对DNA/蛋白质片段进行选择性免疫沉淀,并处理所得片段,分离出DNA和蛋白质成分。
聚合酶链式反应(PCR)、实时PCR、微阵列杂交或直接测序常用来识别限定测序的DNA片段。
ChIP实验可用于研究特异性蛋白质或其修饰同种型与限定基因组区域之间的关联关系。
ChIP正在表观遗传学研究中得到迅速应用。
ChIP实验开始用于在作用机制研究中改进药物研究;以及用于分层医学和诊断;且越来越多地用于ChIP Seq中的DNA测序。
何谓Chromatrap?Chromatrap是一项更有效、更灵敏和更可靠的ChIP方法相比标准方法,Chromatrap®:· 更快· 使用更方便· 更灵敏,对于低丰度目标尤为如此。
Chromatrap®技术更好Chromatrap®套件使用包含一种革新版惰性、多孔聚合物圆盘的离心柱或微孔板,其中蛋白A或蛋白G已与该圆盘共价连接。
这项专利样式独一无二。
在实验期间,染色质/ 抗体复合物选择性地由圆盘保持。
为获得选择性富集DNA,需使用三种缓冲液并按照洗脱步骤洗涤,从而使得Chromatrap®在实验室中更为有效。

染⾊质免疫沉淀技术(ChIP)简介、原理及ChIP ChIP简介:ChIP是染⾊质免疫沉淀技术(Chromatin ImmunoPrecipitation assay)的简称,属于免疫沉淀技术的⼀种,⽤于检测蛋⽩质与DNA的相互作⽤。
染⾊质免疫沉淀技术的⼀个重要⽤途是研究某个转录因⼦A(可以是发⽣某些特定修饰,如磷酸化、⼄酰化等修饰的蛋⽩)是否调控其预期靶基因B的特定转录调控区(主要是启动⼦区域)。
下⾯就以利⽤ChIP研究转录因⼦对基因的调控为例进⾏阐释。
检测⽔平:转录⽔平调控原理及操作流程:染⾊质免疫沉淀技术的原理及⼀般操作流程为:1. 在活细胞状态下,使⽤交联剂(常为甲醛)将蛋⽩质-DNA复合物固定下来;2. 然后通过理化⽅法(常为酶消化法或者超声破碎)将这种复合物中的DNA随机切割为⼀定长度范围内的染⾊质⼩⽚段;3. 继续使⽤蛋⽩质A的特异性抗体I(⼀般要求ChIP级别)处理,将含有蛋⽩质A的蛋⽩质-DNA⽚段特异性标记;4. 再利⽤⼀种可以结合抗体的Protein A(⼀般偶联到分选柱和磁珠上,便于分离),将含有抗体的复合物从作⽤体系中富集分离出来,未被抗体标记的蛋⽩质-DNA则被洗脱去除;5. 将得到的抗体-蛋⽩质-DNA复合物解交联,纯化富集其中的DNA⽚段;6. 利⽤针对⽬的基因B转录调控区的特异性引物(⼀般设计多个位点,覆盖多个区域)进⾏PCR(以前多为半定量PCR,现在随着设备的升级,使⽤荧光定量PCR也逐渐普及)等⼿段检测,如果其中有PCR检出阳性则表明蛋⽩质A可以与基因B的转录调控区有结合(可以是直接也可以是间接结合,具体区分还需要进⾏进⼀步的EMSA检测),⽽具体的结合位点就在引物覆盖区域及其周边位置。
ChIP-on-chip衍⽣技术:ChIP-on-chip有时也称ChIP-chip,要注意其中的⼤⼩写因为它们代表的意义不同,其中前⼀个ChIP表⽰染⾊质免疫沉淀技术,后⼀个chip表⽰基因芯⽚技术。

染色质免疫共沉淀原理测序
染色质免疫共沉淀测序(ChIP-seq)技术原理是:在活细胞状态下固定蛋白质-DNA复合物,并将其随机切断为一定长度范围内的染色质小片段,然后通过免疫学方法沉淀此复合体,特异性地富集目的蛋白结合的DNA片段,通过对目的片断的纯化与检测,从而获得蛋白质与DNA相互作用的信息。
ChIP-seq技术进一步对富集得到的DNA片段进行高通量测序,是目前在全基因组水平研究蛋白结合靶DNA序列的重要手段,为转录因子、组蛋白修饰、核小体定位等表观遗传学的研究提供有效方法。
以上内容仅供参考,如需获取更多详细信息,建议咨询生物学专家或查阅染色质免疫共沉淀测序相关书籍文献。

ChIP试剂盒染色质免疫共沉淀全套解决方案染色质免疫沉淀-芯片试剂盒染色质免疫沉淀(芯片)的完整溶液是研究体内DNA与蛋白质相互作用的最佳方法。
其基本原理是将蛋白质-DNA复合物固定在活细胞状态,将其随机切割成一定长度范围内的小染色质片段,然后通过免疫学方法沉淀复合物,特异性富集与靶蛋白结合的DNA片段。
通过目标片段的纯化和检测,可以获得关于蛋白质和DNA之间相互作用的信息。
芯片不仅可以检测体内反式因子与DNA的动态相互作用,还可以研究组蛋白的各种共价修饰与基因表达的关系。
此外,芯片与其他方法的结合扩大了其应用范围:通过芯片与基因芯片的结合建立的芯片-芯片法已广泛用于高通量筛选特异的反式因子靶基因;芯片结合体内足迹法寻找反式因子的体内结合位点;核糖核酸芯片用于研究核糖核酸在基因表达调控中的作用因此,随着ChIP的进一步完善,它必将在基因表达调控研究中发挥越来越重要的作用。
就目前国内研究状况而言,教师在研究领域有分化、转录、发育、诱导多能性、肿瘤干细胞、表观遗传学等。
会做ChIP实验,一些老师会自己购买抗体,手动配置试剂。
然而,因为实验本身具有复杂的实验步骤,并且其中许多步骤非常关键,需要更多的试剂,所以很容易导致配置之间的错误,并且实验周期长。
如果没有设置阴性和阳性对照,结果就无法分析,从而导致无休止的混乱。
经典染色质免疫沉淀(ChIP)试剂盒(p-2002):提供细胞样品上染色质免疫沉淀反应所需的所有试剂此外,试剂盒包含阳性对照抗体(核糖核酸聚合酶2抗体)、阴性对照抗体(正常小鼠的IgG)和GAPDH引物(可用作阳性对照,以确保试剂盒中的试剂和操作步骤没有问题)在大多数生长中的哺乳动物细胞中,核酸聚合酶II富集在GAPDH基因启动子上,为启动转录做准备,因此启动子可以与核酸聚合酶II进行免疫沉淀反应,但不能与正常的小鼠IgG进行免疫沉淀反应。
在该染色质免疫沉淀反应中,细胞与甲醛偶联以提取其中的染色质染色质被适当破坏,然后加入微孔中,与吸附在微孔表面的抗体反应特异性结合在微孔上的DNA从抗体-捕获蛋白-DNA复合物中释放出来,通过我们公司专门设计的高速离心柱进行翻转和纯化。

实验染色质免疫沉淀技术(ChIP)实验操作指南全心全意就为医生服务,一心一意只为造福医生。
真核生物的基因组DNA以染色质的形式存在。
因此,研究蛋白质与DNA在染色质环境下的相互作用是阐明真核生物基因表达机制的基本途径。
染色质免疫沉淀技术(chromatin immunoprecipitation assay, CHIP)是目前最常用研究体内DNA与蛋白质相互作用的方法。
它的基本原理是在活细胞状态下固定蛋白质-DNA复合物,并将其随机切断为一定长度范围内的染色质小片段,然后通过免疫学方法沉淀此复合体,特异性地富集目的蛋白结合的DNA片段,通过对目的片断的纯化与检测,从而获得蛋白质与DNA相互作用的信息。
CHIP不仅可以检测体内反式因子与DNA的动态作用,还可以用来研究组蛋白的各种共价修饰与基因表达的关系。
而且,CHIP与其他方法的结合,扩大了其应用范围:CHIP与基因芯片相结合建立的CHIP-on-chip方法已广泛用于特定反式因子靶基因的高通量筛选;CHIP与体内足迹法相结合,用于寻找反式因子的体内结合位点;RNA-CHIP用于研究RNA 在基因表达调控中的作用。
由此可见,随着CHIP的进一步完善,它必将会在基因表达调控研究中发挥越来越重要的作用。
实验准备实验材料:细胞样品试剂、试剂盒:甲醛、甘氨酸、PBS、SDS Lysis Buffer、洗脱液、RNaseA、蛋白酶K、omega胶回收试剂盒仪器、耗材:离心管、超声仪、电泳仪、离心机ChIP的一般流程甲醛处理细胞→ 收集细胞,超声破碎→ 加入目的蛋白的抗体,与靶蛋白-DNA复合物相互结合→ 加入ProteinA,结合抗体-靶蛋白-DNA复合物,并沉淀→ 对沉淀下来的复合物进行清洗,除去一些非特异性结合→ 洗脱,得到富集的靶蛋白-DNA复合物→ 解交联,纯化富集的DNA-片断→ PCR分析。
请ChIp具体操作流程:第一天(一)、细胞的甲醛交联与超声破碎1、取出1平皿细胞(10cm平皿),加入243 μl 37%甲醛,使得甲醛的终浓度为1%(培养基共有9ml)。

Chromatin Immunoprecipitation (ChIP) Analysis for Protein-DNA InteractionsWeike Si, 6/22/05; Commented by TCH 8/6/05General Background and ConsiderationsChIP is a widely used method to identify specific proteins associated with aregion of the genome, or in reverse, to identify regions of the genomeassociated with specific proteins. These proteins can be isoforms ofhistones modified at a particular amino acid or other chromatin associatedproteins. When employed with antibodies that recognize histonemodifications, ChIP can be used to “measure” the amount of themodification. An example of this would include measurement of the amountof histone H3 acetylation associated with a specific gene promoter regionunder various conditions that might alter expression of the gene. Histonesare not the only proteins that can be studied using this technique. Much ofthe recent interest has been in analyzing transcription factor distributionthroughout the genome or at specific loci.When performing ChIP, cells are first fixed with formaldehyde tocrosslink proteins to DNA and then chromatin is harvested from the cellsand subjected to an immunoselection process, which requires the use ofspecific antibodies. Any DNA sequences cross-linked to the protein ofinterest will co-precipitate as part of the chromatin complex. After theimmunoselection of chromatin fragments and purification of associatedDNA, the detection of specific DNA sequences is performed. If the DNAwhich will be detected is associated with the protein or histone modificationbeing examined, the relative representation of that DNA sequence will beincreased (or enriched) by the immunoprecipitation process.Generally, standard PCR is performed to identify the DNA sequence(the gene or region of the genome) associated with the protein of interest.The relative abundance of a specific DNA sequence isolated via theprotein-specific immunoselection is compared to DNA obtained when usingan unrelated antibody control. DNA fragments are run on gels to facilitate quantitation of the PCR products. A much more accurate alternative to standard PCR is real time quantitative PCR (RT-qPCR). Cloning of sequences from a ChIP experiment is also possible, to create libraries of fragments that are enriched for those that interact with a particular protein. The combination of chromatin IP with microarray applications (ChIP on chip) is a novel technique that is becoming more popular, allowing the generation of genome-wide maps of protein-DNA interactions or histone modifications.References:Das PM, et al., Biotechniques 37: 961-969, 2004Luo, R.X., et al., Cell 92: 463-473, 1998.Braunstein, M., et al., Mol. Cell. Biol. 16: 4349-4356, 1996.Manabe, I., et al., J. Clin. Invest. 107: 823-834, 2001.Cervoni, N., & Szyf, M., J. Biol. Chem. 276: 40778-40787, 2001.Materials and ReagentshiTE Buffer: 50mM Tris-HCl, 10mM EDTA, pH 7.5.PBS: 10mM Na2HPO4 (dibasic, anhydrous), 2mM KH2PO4 (monobasic, anhydrous), pH 7.4, 150mM NaCl, 10mM sodium phosphate.Formaldehyde: from Fisher ScientificChIP Lysis Buffer: 50mM HEPES/KOH pH 7.5, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate. Add protease inhibitors prior to use.High Salt ChIP Wash Buffer: 50mM HEPES, pH 7.5 by KOH, 500mM NaCl, 1mM EDTA, 1% Triton X-100,0.1% SDS, 0.1% sodium deoxycholate. Add protease inhibitors prior to use. Protease Inhibitor Cocktail: Complete Protease Inhibitor Cocktail tablets (Roche Biochemicals). One tablet per 10ml of PBS or Lysis Buffer.ChIP Elution Buffer: hiTE Buffer containing 1% SDS.Protein A/G agarose: Sepharose 4B Protein G beads (GE Health Amersham Pharmacia). Glycine: 2.5M Glycine.Sonicator: Fisher Scientific F60 Model5M NaClPart I. Optimization of DNA ShearingOne of the important parameters for ChIP assay is to establish optimal conditions required for shearing cross-linked DNA to 200-1000 base pairs in length. Optimal conditions required for shearing cross-linked DNA to 200-1000 base pairs in length depend on the cell type, cell concentration per lysis buffer and thesonicator equipment, including the power settings and number of pulses.We are using Fisher’s F60 Sonicator and our experience shows DNA is sheared to theappropriate length with 12-second pulses x 4-5 (i.e., keep 1~2min. between pulses; makesure samples are on ice all times)at 80% of maximum power (i.e., Setting = 14). Oncesonication conditions have been optimized, keep cell number consistent for subsequentexperiments. You’re strongly encouraged to optimize the DNA shearing condition using thefollowing two methods. Method #1 is simplistic and should be initially used to obtain thepreliminary settings for further testing described in Method #2. If you have already obtainedsome of the sonication conditions, you can directly proceed with Method #2.Be sure to keep the sample on ice at all times as the sonication generates heat whichwill denature the DNA and proteins. Check the size of sonicated DNA by gelelectrophoresis after reversion of cross-links.Method #1: Direct use of purified genomic DNA.1. Add 1~2ug of purified genomic DNA into the 2-5ml microfuge tubes (the # of tubes willdepend on how many sonication conditions you want to test). Bring up the volume in each tube to 200ul with ddH2O, Keep samples on ice.2. Perform sonication (Fisher’s F60 model) by changing either the power settings (e.g., fullscale = 20; 80% = 16; and 70% = 14) and/or the number of 10 to 15-second pulses(usually between 3 to 5 times). Please keep samples on ice all time; wait for 1-2 min.between pulses to avoid rapid heating up of the samples.3. Transfer the sonicated DNA samples to a fresh set of 1.7ml tubes. Perform ethanolprecipitation using our regular lab protocol.4. Resuspend the DNA samples in 10-20ul of ddH2O. Load onto the1.2% ~ 1.5% agaroseminigel and run it for 40-60min at 60-70 volts. Ideally, the center of the DNA smearshould migrate along the 500bp-position.Method #2: Use of the cross-linked cells.Note: Whenever possible, place samples on ice.1. Plate C3H10 cells in one T-75 flask (in 20ml complete medium) at 70% confluency at37C 5% CO2 incubator. It should reach 80-90% confluency overnight, yielding ~1 x 107 cells.2.Crosslink proteins to DNA by adding 540ul of 37% Formaldehyde directly to the 20mlcells culture medium (at a final concentration of 1% Formaldehyde). Incubate the flask room temperature for 5-10 minutes.3. Add 1.0ml of 2.5M Glycine (final concentration 125 mM) to the medium for 10min, at roomtemperature to quench the formaldehyde.4. Aspirate medium, removing as much medium as possible. Wash cells using 5 ml of icecold PBS containing protease inhibitors (i.e., we are using Roche’s Complete PI cocktail tablets). Note: Add protease inhibitors to PBS just prior to use.5. Add 2ml cold PBS, and scrape cells into a 50-ml conical tube.6. Pellet cells for 3000 rpm, 5 minutes at 4ºC. Remove PBS and add 2.0 ml ChIP LysisBuffer containing protease inhibitors to lyse cells for 30 min on ice.7. Prepare multiple 200µl aliquots for sonication. Note: The 200 ul of ChIP Lysis Buffer is per 2 X 106cells; if more cells are used, the resuspended cell pellet should be divided into 200µl aliquots so that each 200µl aliquot contains ~1 X 106 cells.8. Sonicate lysate to shear DNA to lengths between 200 and 1000bp being sure to keepsamples ice cold (e.g., 80% power ×12sec×4times, between pulses incubate on ice for 1-2min).9. Recover DNA by phenol/chloroform extraction, ethanol precipitation, wash ×2. Runsamples (e.g., 20ul per sample) in 1.5% agarose gel to visualize shearing efficiency.Part II. Chromatin Immunoprecipitation ProtocolNote: Numerous controls can be set up. Most common ones are treated (e.g., AdWnt3A) vs.untreated (e.g., AdGFP), and/or gene-specific antibody (e.g., anti-β-catenin) vs.non-specific antiserum (e.g., a control IgG). Additionally, PCR reactions can be carried out to detect control genomic loci (e.g., GAPDH promoter region). A. In Vivo Crosslinking and LysisPrior to starting this section:•Obtain ice for incubation of PBS (see Step 3) and for incubating culture dish (see Step 6).•Prepare 1X PBS and put on ice. This will be used for washes and needs to be ice cold.•Warm SDS Lysis Buffer to room temperature to ensure SDS is in solution before proceeding with cell lysis.1. Plate C3H10 T1/2 cells in T-75 flasks at 70-80% confluency.2. Infect cells with an optimal titer of AdWnt3A or AdGFP in T-75 flasks containing 20ml ofgrowth media. For C3H10T1/2 cells, there are approximately 1 x 107 cells per T-75flask. This will generate a preparation of chromatin that can be used for up to 5 separate immunoprecipitations per flask.3. At 36 to 45hrs after infection, add 540µl of 37% Formaldehyde to 20ml of growth media(Final concentration is ~1%) to crosslink and gently swirl the flask to mix.4. Incubate at room temperature for 10 minutes.10m l of ice cold 1X PBS to a separate tube for every T-75 flask and remove5. Meanwhile,add one tablet of the Complete PI tablet. Put on ice.6. Add1.0ml of2.5M Glycine (final concentration 125 mM) to each T-75 flask to quenchexcessive Formaldehyde.7. Swirl to mix and incubate at room temperature for 5 minutes.8. Aspirate medium as completely as possible, being careful not to disturb the cells.10ml of cold 1X PBS to wash cells.9. Add10. Remove PBS. Add 1.0ml cold PBS containing Protease Inhibitor Cocktail. Scrape cells from each flask into a microfuge tube. Spin at 700xg at 4°C for <5 min. to pellet cells. 11. During spin, prepare ~10ml of ChIP Lysis Buffer containing Complete PI InhibitorsCocktail .12. Resuspend cell pellet in 1.0ml of ChIP Lysis Buffer with PI cocktail. Cell Density isimportant for reliable cell lysis. Adjust accordingly if different cell concentrations aredesired as the ratio of lysis buffer to cell (for every 1x107 C3H10T1/2 cells,1.0ml of Lysis Buffer is recommended for this protocol).13. Aliquot between 200µl per microfuge tube. Lysate can be frozen at -80°C at this step.14. If optimal conditions for sonication have already been determined, proceed to Section B.B. Sonication to Shear DNAPrior to starting this section:Optimal conditions required for shearing cross-linked DNA to 200-1000bps in length need to be determined as described in Part I.1. If desired, remove 5µl of cell lysate from Section A, Step 13 for agarose gel analysis ofunsheared DNA. If lysate from Section A, Step 13 was previously frozen, thaw on ice.2. Sonicate cell lysate on ice using the conditions optimized in Part 1, Method #2. Shearedcross-linked chromatin can be stored at -80°C for up to a few months.3. Spin at 12,000-15,000 x g at 4°C for 10 minutes to remove insoluble material,transfersupernatant into new sterile microtubes in 100µl aliquots. Each 100µl aliquot contains 2 x 106 cell equivalents of lysate which is enough for one immunoprecipitation.C. Immunoprecipitation (IP) of Crosslinked Protein/DNAenough ChIP Lysis Buffer containing protease inhibitors for the number of1. Preparedesired immunoprecipitations and store on ice.• Each IP requires the addition of 900µl of ChIP Lysis Buffer with PIs.• Samples include Wnt3A vs. GFP-treated; anti-β-catenin vs. mouse IgG control. Insome cases, no antibody/IgG controls can also be set up.2. Prepare one microfuge tube containing 100µl of sheared crosslinked chromatin (SectionB, step 3) for the number of desired immunoprecipitations and put on ice. If lysate has been previously frozen, thaw on ice.• Alternatively, if multiple immunoprecipitations will be performed from the same lysate preparation, place the entire volume for the number of desired immunoprecipitations in one large tube that will be able to accommodate a volume of 1.1ml for each IP.• Each 100µl will contain ~2 x 106 cell equivalents of chromatin.400µl of ChIP Lysis Buffer into each tube containing 100µl of cell lysate/chromatin.3. Add4. Add60µl of pre-washed Protein G Agarose for each IP.• The Protein G Agarose is 50% slurry, which should be washed in ChIP Lysis Buffer containing PI cocktails twice. Gently mix by inversion before removing.• This step serves to “preclear” the chromatin, i.e., to remove proteins or DNA that may bind nonspecifically to the Protein G agarose.5. Incubate for 1 hour at 4°C with rotation.6. Pellet agarose by brief centrifugation (3000-5000 x g for 1 minute).• Do not spin Protein G Agarose beads at high speeds. Applying excessive g-force may crush or deform the beads and cause them to pellet inconsistently.10µl (1%) of the supernatant as Input and save at 4°C until Section D, step 1.7. Remove• If different chromatin preparations are being carried together through this protocol,remove 1% of the chromatin as Input from each.supernatant by aliquoting 400-500 µl into fresh microfuge tubes.the8. Collect9. Add the immunoprecipitating antibody to the supernatant fraction:• For anti-β-catenin, add 1.0-10µg of antibody per tube.• For the negative control, Normal Mouse IgG, add 1.0µg of antibody per tube.10. Incubate at 4°C for 2~4 hours with rotation.• It may be possible to reduce the incubation time of the IP. This depends on manyfactors (antibody, gene target, cell type, etc.) and will have to be tested empirically. 11. Add 100µl of Protein G Agarose (pre-washed with ChIP Lysis Buffer) for 1 hour at4°C with rotation.• This serves to collect the antibody/antigen/DNA complex.12. Pellet Protein G Agarose by brief centrifugation (3000-5000 x g for 1 minute) andremove the supernatant fraction.13. Wash the Protein G Agarose-antibody/chromatin complex by resuspending the beads in1.0ml each of the cold buffers containing PI cocktail in the order listed below andincubating for 3-5 minutes on a rotating platform followed by brief centrifugation (3000-5000 x g for 1 minute) and careful removal of the supernatant fraction:a) ChIP Lysis Buffer wash 5min × 2 times, at RTb) High Salt ChIP Lysis Buffer (w/0.5M NaCl) wash 5min × 2 times, at RTc) ChIP Lysis Buffer wash 5min × 1 times, at RTd) hiTE buffer wash 5min × 1 times, at RTD. Elution of Protein/DNA ComplexesPrior to starting this section:• Bring ChIP Elution Buffer to room temperature. A precipitate may be observed but will go into solution once room temperature is achieved.• Set water bath to 65°C for use in Section E.1. MakeChIP Elution Buffer for all IP tubes and all Input tubes (see Section C, step 7).Input tubes (see Section C, step 7), add 200µl of Elution Buffer and set aside at 2. Forroom temperature until Section E.100µl of Elution Buffer to each tube containing the antibody/agarose complex. Mix 3. Addby flicking tube gently.4. Incubate at room temperature for 15 minutes.5. Pellet agarose by brief centrifugation (3000-5000 x g for 1 minute) and collectsupernatant into new microfuge tubes.6. Repeat steps 4-6 and combine eluates (total volume = 200µl).E. Reverse Crosslinks of Protein/DNA Complexes to Free DNA1. To all tubes (IPs and Inputs) add 8µl of 5M NaCl and incubate at 65°C for 4-5 hours (orovernight) to reverse the DNA – Protein crosslinks (Note: This step can also becarried out in PCR heating blocks). After this step the sample can be stored at -20°C and the protocol continued the next day.2. Optional: to all tubes, add 1µl of RNase A and incubate for 30 minutes at 37°C.3. Optional: add 4µl 0.5M EDTA, 8µl 1M Tris-HCl and 1µl Proteinase K and incubate at45°C for 1-2 hours.F. DNA Purification and Quantitative Real-Time PCR Analysis1. To each uncrosslinked sample (~200µl), add 100µl of 7.5M (NH4)2OAc and 250µl ofPC-8. Perform PC-8 extraction as our regular lab protocol. Repeat PC-8 extraction(s) if necessary.2. To each sample, add 5µl seeDNA co-precipitate to each sample and mix well. Add700µl cold 100% ethanol. Spin samples (in 1.7ml Eppendorf tubes) at top speed for 5 min. Wash pellets with 70% ethanol twice. Air-dry the pellets.3. Dissolve samples in 100µl ddH2O. Samples are ready for being used for real-time PCR/regular PCR analysis or Kept at -20C or -80C.PCR /regular PCR use our regular lab protocols.4. Performreal-time。

染色质免疫沉淀技术介绍染色质免疫沉淀技术(Chromatin Immunoprecipitation,ChIP)是一种用于研究染色质上特定蛋白质与DNA的相互作用的实验方法。
通过该技术,我们可以确定某个蛋白质在染色质上的结合位点,进而探究基因表达调控、表观遗传学和疾病发生等重要生物学问题。
ChIP的原理ChIP技术的基本原理是利用特异性抗体与目标蛋白质结合,然后通过免疫沉淀的方式将蛋白质及其结合的DNA分离出来。
具体步骤如下:1. 交联首先,将细胞或组织进行交联,使得染色质上的蛋白质与DNA形成稳定的结合。
常用的交联剂包括甲醛和二氧化硅。
2. 细胞裂解将交联后的细胞或组织进行裂解,释放出染色质。
3. DNA切割使用限制性核酸内切酶或超声波等方法将染色质切割成小片段。
切割后的DNA片段长度通常在200-1000碱基对之间。
4. 免疫沉淀将特异性抗体与目标蛋白质结合,形成抗原-抗体复合物。
然后将抗原-抗体复合物与染色质中的目标蛋白质结合的DNA片段一起免疫沉淀。
5. 分离DNA通过洗涤等步骤将非特异性结合的DNA片段去除,保留与目标蛋白质结合的DNA片段。
6. 解交联去除染色质与蛋白质的交联,使得DNA片段恢复单链状态。
7. DNA纯化将解交联后的DNA片段进行纯化,去除杂质。
8. DNA分析通过PCR、测序等方法对免疫沉淀得到的DNA片段进行分析,确定目标蛋白质结合的DNA序列。
应用ChIP技术在生命科学研究中得到了广泛应用,尤其是在以下领域:1. 基因表达调控通过ChIP技术,可以确定转录因子与染色质上的结合位点,进而揭示基因的调控机制。
研究人员可以通过ChIP-Seq等方法,高通量地鉴定转录因子结合位点,从而识别出与特定基因调控相关的转录因子。
2. 表观遗传学ChIP技术可以用于研究染色质修饰与基因表达调控之间的关系。
例如,通过ChIP-Seq可以鉴定出与DNA甲基化和组蛋白修饰相关的位点,进一步探究这些修饰与表观遗传学调控的机制。
染色质免疫共沉淀ChIP中文操作流程1.细胞中加入1%的甲醛,8ml的培养液加入216 ul的甲醛,37度十分钟。
2.配制含有蛋白酶抑制剂的PBS 20 ml和含有蛋白酶抑制剂的SDS溶液1ml3.将细胞拿出来,迅速的移除含甲醛的培养基,加入含蛋白酶抑制剂的PBS洗两遍。
胰酶消化20秒,加入含蛋白酶抑制剂的PBS 1ml。
用细胞刮刀把细胞刮下,收集到1.5ml 的离心管里面。
4.4度2000rpm离心10min,弃上清液,加入200ul含蛋白酶抑制剂的SDS溶液。
吹打重悬细胞,冰上孵育10分钟。
5.超声切割DNA,总切割时间4min30sec,超声10sec,间隙10sec。
6.4度13000rpm离心10min,转移上清液到一个新的2ml的离心管,弃沉淀。
7.稀释超声后的上清液到10X的CHIP稀释液,200ul的上清液加入1.8ml的CHIP稀释液,达到最终体积2ml。
8.为去除非特异性,加入75ul的Salmon Sperm DNA/Protein A Agarose-50% Slurry,4度旋转30分钟。
9.1000rpm离心3min沉淀Salmon Sperm DNA/Protein A Agarose-50% Slurry,收集上清液。
10.上清液加入1抗,4度振荡过夜。
11.加60ul的Salmon Sperm DNA/Protein A Agarose-50% Slurry,沉淀抗体/抗原复合物,4度旋转一小时。
12.1000rpm 4度3min收集沉底,移除上清液,开始洗脱过程。
13.低盐免疫复合物洗脱液,旋转5min,1000rpm离心3min收集沉淀14.高盐免疫复合物洗脱液,旋转5min,1000rpm离心3min收集沉淀15.Licl免疫复合物洗脱液,旋转5min,1000rpm离心3min收集沉淀16.TE Buffer,旋转5min,1000rpm离心3min收集沉淀,两次17.现在得到的是protein A/antibody/histone/DNA complex,新制备elution buffer (1%SDS,0.1M NaHCO3)。
染色质免疫共沉淀原理
染色质免疫共沉淀(ChIP)是一种用于研究蛋白质与DNA相互作用的技术。
该技术利用抗体特异性地结合目标蛋白质,然后通过共沉淀的方式将与该蛋白质结合的DNA分离出来,从而研究蛋白质与DNA的相互作用。
在染色质免疫共沉淀技术中,首先需要对目标蛋白质进行抗体的制备。
然后将细胞或组织样品进行交联,使得蛋白质与DNA之间的相互作用得以保留。
接着,将交联后的样品进行裂解,使得细胞核内的DNA暴露出来。
然后,将抗体与样品混合,使得抗体能够特异性地结合目标蛋白质。
随后,将混合物进行共沉淀,使得与目标蛋白质结合的DNA能够被分离出来。
最后,通过PCR、测序等方法对分离出的DNA进行分析,从而研究蛋白质与DNA的相互作用。
染色质免疫共沉淀技术在生物学研究中有着广泛的应用。
例如,该技术可以用于研究转录因子与DNA的相互作用,从而揭示基因调控的机制。
此外,该技术还可以用于研究组蛋白修饰与DNA的相互作用,从而揭示染色质结构与功能的关系。
此外,该技术还可以用于研究病原微生物与宿主细胞的相互作用,从而揭示病原微生物的致病机制。
染色质免疫共沉淀技术是一种重要的生物学研究技术,可以用于研究蛋白质与DNA的相互作用,从而揭示生物学过程的机制。
染色质免疫共沉淀结果解析
染色质免疫共沉淀(ChIP)是一种常用的分子生物学技术,用于检测特定蛋白质与基因组DNA的相互作用。
通过该技术,可以确定某种特定的蛋白质是否与某一特定的DNA序列结合,并能够分析这种结合的模式和位置。
ChIP的实验步骤大致分为以下几步:交联、裂解、抗体免疫沉淀、洗涤和提取。
其中,交联是将细胞中的蛋白质与DNA“固定”在一起,裂解则是将细胞核内的染色质分解成小碎片以便于后续的操作。
抗体免疫沉淀是利用特定的抗体将要检测的蛋白质与DNA结合物质
免疫沉淀出来,洗涤则是将非特异性的蛋白质和DNA从结合物质中洗去,提取则是将免疫沉淀得到的物质提取出来以便于后续的分析。
对于ChIP实验的结果解析,需要进行数据处理和分析。
最常用
的方法是将ChIP所得的DNA片段进行PCR扩增,然后进行基因测序
和比对分析。
通过对比对结果的分析,可以确定特定的蛋白质与DNA 序列的结合情况,并确定它们的相互作用模式和位置。
另外,还可以利用一些计算机软件如MACS和HOMER等进行数据处理和分析,以及
进行统计学分析和可视化展示。
综上所述,染色质免疫共沉淀技术是一种重要的分子生物学技术,能够帮助我们了解蛋白质与DNA相互作用的模式和位置,从而为后续的基因功能研究和临床诊断提供重要的参考依据。
- 1 -。
染色质免疫共沉淀测序
1质粒染色质免疫共沉淀测序
质粒染色质免疫共沉淀测序(ChIP-seq)是近年来非常受欢迎的基因组学技术,它使用免疫沉淀技术研究基因的表达。
质粒染色质免疫共沉淀是一种高通量的技术,可以节省时间和金钱,也可以快速有效地定位和定性识别染色质位点。
它在研究染色质及其表达调控相关位点时有着重要使用。
ChIP-seq将组织中的染色质细胞分离、捕获特定结合到特定蛋白质上的片段,并使用高通量测序技术来鉴定相应的序列。
在染色质投射过程中,基因组DNA从染色质上经过免疫沉淀(IP)和PCR扩增,以及液相芯片的方式鉴定某一特定的DNA片段。
ChIP-seq过程需要从每个步骤中取得高质量的数据,这就要求研究人员在实验室操作上具有良好的能力。
基于ChIP-seq确定依赖染色质结合位点可以有效地鉴定和定性识别调节因子结合对基因表达的影响,从而研究染色质结合部位的功能特征。
此外,ChIP-seq技术还可以检测某种基因类型的调控行为,如转录因子(TF)结合到基因组中的位点,从而研究其表达调控和功能及其相关分子,如DNA修饰或蛋白质-蛋白质相互作用。
因此,质粒染色质免疫共沉淀测序对于研究基因有着重要的意义,是当今分子生物学领域非常热门的技术。
ChIP-seq不仅可以为基因组项目提供有效的数据,还可以节省实验室操作的成本和时间。
染色质免疫共沉淀内参基因染色质免疫共沉淀(ChIP)是用来研究蛋白质与染色质相互作用的一种实验技术,通过利用特异性抗体来富集与目标蛋白质结合的染色质片段。
然而,在进行ChIP实验时,需要使用内参基因来进行标准化和校正实验结果,以确保实验的准确性和可靠性。
内参基因是指在特定条件下表达稳定、不受外界因素干扰的基因。
在ChIP实验中,内参基因被用来标准化目标基因的富集水平,以消除实验中可能出现的误差。
下面将从内参基因选择、内参基因验证及常用内参基因几个方面来详细探讨。
一、内参基因选择选择适当的内参基因是进行ChIP实验的关键步骤之一。
一个理想的内参基因应具备以下特点:1. 稳定的表达水平:内参基因的表达水平应在不同样品、不同处理条件下保持稳定,不受干扰因素的影响。
这可以通过实时定量PCR (qPCR)或基因芯片技术来评估基因的表达稳定性。
2. 细胞类型特异性:内参基因的表达水平应在所研究的细胞类型中具有一定特异性,并且不受目标蛋白质结合与浓度的影响。
这可以通过在不同细胞类型中进行实时定量PCR检测来评估。
3. 控制组条件下表达水平不变:在ChIP实验中,常常需要对比不同样品或处理条件下的富集水平。
因此,内参基因的表达水平应在不同组别条件下保持相对稳定,以确保实验结果的可比较性。
根据以上的特点,常用的内参基因包括GAPDH、ACTB、GUSB等,这些基因的表达水平通常在不同细胞类型和处理条件下比较稳定,且与ChIP实验中的目标基因的结合无关。
二、内参基因验证为确保选择的内参基因在实验条件下满足稳定性和特异性等要求,进行内参基因验证是必要的。
验证内参基因的常用方法有:1. 实时定量PCR:通过实时定量PCR测定一系列候选内参基因的表达水平,根据表达的稳定性和特异性选择最合适的内参基因。
在实验中,可以根据ChIP-qPCR的结果及对应内参基因的表达情况,评估其在ChIP实验中的可靠性。
2. 基因芯片技术:利用基因芯片技术可以同时检测大量基因的表达水平,对于内参基因的选择具有更高的精确性和鉴定能力。