无菌制剂风险管理
- 格式:ppt
- 大小:262.50 KB
- 文档页数:45


《药品GMP指南》—无菌药品
质量风险管理
一、法规要求
二、质量风险点
质量风险管理的两大主要原则:质量风险的评估应基于科学知识并与保护患者相联系;质量
风险管理的资源投入、形式和文件应与风险的水平相适应。
无菌制剂是指法定药品标准列有无菌检查项目的制剂。
注射剂指药物制成的供注入人体内的无菌溶液(包括乳浊液和混悬液)以及供临用前配成溶液或混悬液的无菌粉末或浓溶液。其质量保证的重点在于无菌保证、细菌内毒素和微粒污染的控制。也需要特别关注防止混淆和交叉污染。
三、最终灭菌产品的质量风险评估案例
(一)无菌保证风险与质量风险控制点
无菌保证的风险主要来自于:产品灭菌前微生物污染水平;灭菌工艺的可靠性;容器密封的完整性;无菌保证管理体系。
1.产品灭菌前微生物污染水平
产品灭菌前通常都存在一定程度的微生物。微生物污染主要受以下因素影响:
原材料和包装材料中的微生物——风险在于其可能进入产品。质量风险控制方法:
制定原辅料采购标准,规定微生物限度。通常应不超过100CFU/g,并不得含致病菌。
进行供应商的确认时应重点关注供应商的生产过程对微生物污染、细菌内毒素的污染、产品混淆和交叉污染风险的控制措施。
对供应商及其供应的原料进行年度质量回顾分析,以评估其质量状况。对不良趋势的供应商应采取针对性的措施,如增加现场检查的频率,更加严格的抽样方案。
严格管理仓储条件,确保原料储存过程中质量受控。如干燥、防虫、防鼠等。包装材料如玻璃瓶应定点采购,其包装应能防止昆虫进入,储存过程防止受潮长霉。
2.生产环境
注射剂的生产从原料称量开始直至完成密封,都分别在相应的卫生洁净区中进行。生产过程各步骤都可能存在药物直接暴露于环境的环节,存在来自于生产环境的微生物污染的风险。如果能证明在动态下生产区的洁净度能符合上述标准,则来自环境的微生物污染风险是较低的。
3.生产设备
注射剂通常采用固定的设备,安装了生产及在线清洗、消毒等多种工艺管道。因此设备存在残留物或微生物的可能,对产品有潜在风险。质量风险控制方法:

无菌制剂生产质量管理规范
2004年9月 美国FDA发布
2009年6月 药审中心组织翻译
武田药品有限公司翻译
北核协会审核
药审中心最终核准
目 录
Ⅰ. 前言.........................................................................................................................1
Ⅱ. 背景.........................................................................................................................1
A. 规章制度...............................................................................................................1
B. 技术框架...............................................................................................................2
Ⅲ. 范围.........................................................................................................................2
Ⅳ. 厂房和设施.............................................................................................................3
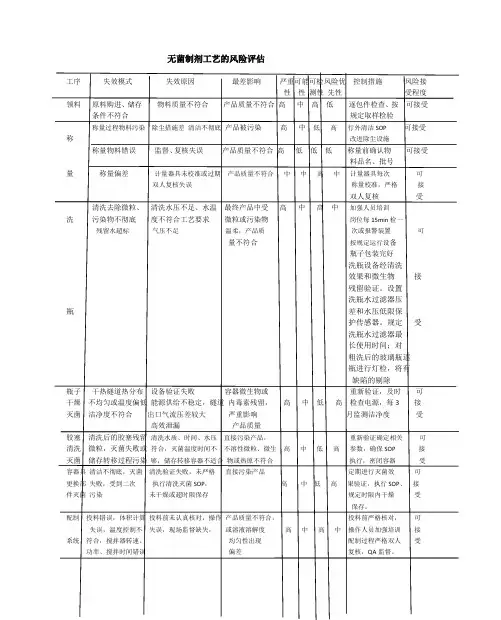
无菌制剂工艺的风险评估
工序 失效模式 失效原因 最差影响 严重可能可检风险优 控制措施 风险接
性 性 测性 先性 受程度
领料 原料购进、储存 物料质量不符合 产品质量不符合 高 中 高 低 逐包件检查、按 可接受
条件不符合 规定取样检验
称量过程物料污染 除尘措施差 清洁不彻底 产品被污染 高 中 低 高 行外清洁SOP 可接受
称 改进除尘设施
称量物料错误 监督、复核失误 产品质量不符合 高 低 低 低 称量前确认物 可接受
料品名、批号
量 称量偏差 计量器具未校准或过期 产品质量不符合 中 中 高 中 计量器具每次 可
双人复核失误 称量校准,严格 接

Part
1、引言
2010年版《药品生产质量管理规范》提出了质量风险管理的要求,应当根据科学知识及经验对质量风险进行评估,以保证产品质量。
此外,根据GMP第四十六条第一款要求:应当综合考虑药品的特性、工艺和预定用途等因素,确定厂房、生产设施和设备多产品共用的可行性,并有相应评估报告。
针对我国目前药品生产企业存在的多产品共线、生产设施、系统、设备共用的问题,ISPE基准指南7《基于风险分析的制药产品生产》提出了“初始暴露原理”这一观点。
初始暴露原理,即暴露/交叉污染如何产生的,应考虑以下四个基本因素:
(1)混淆,即错误的物料;
(2)清洁(残留),不充分的清洁;
(3)机械转移过程,将残留风险从一件产品转移到另一产品;
(4)空气转移,即空气中的粉尘接触产品、设备。这四个基本因素归纳起来便是混淆与交叉污染。
因此,本文将从混淆与交叉污染两个层面,对无菌粉针剂产品共线生产的风险进行评估与分析。
Part
2、无菌粉针剂产品共线生产风险评估的目的
根据无菌粉针剂产品的特性、工艺和预定用途等因素,无菌粉针剂产品共线生产风险评估的目的是:评估多产品无菌制剂生产使用的厂房、设施及设备共用的可行性,对多品种共线生产可能发生混淆、污染与交叉污染的风险点进行识别,提出降低混淆、污染与交叉污染风险的措施,并加以文件标准化。
另外,因根据风险评估的结果,指导药品生产活动的相关验证。
Part
3、无菌粉针剂产品共线生产风险评估与分析
3.1
产品特性的风险分析
根据《药品生产质量管理规范》要求,对于生产特殊性质的药品,如高致敏性药品(如青霉素无菌制剂),必须采用专用和独立的厂房、生产设施和设备;生产β-内酰胺结构类的无菌制剂,必须使用专用设施(如独立的空气净化系统)和设备,并与其他药品生产区严格分开。
对于共线生产的无菌药品,应对它们的产品特性进行分析和识别,从产品的剂型、性状、活性成分、水中溶解度、LD50、致敏性以及共线生产产品之间是否有直接或间接的配伍禁忌等多方面综合评估,选择溶解度差、毒性及致敏性强的产品进行清洁验证,采用合适的清洁方法,防止设备、零部件、取样工具等之间的交叉污染。对于存在配伍禁忌的药品,可考虑采用单独的配液系统、灌装(分装)零部件,在清洁程序结束后应进行必要的检验,符合要求后才能供其他产品生产使用。