(整理)当代有机氟化学
- 格式:doc
- 大小:8.10 MB
- 文档页数:45

第二章:高分子材料概论2.9有机氟材料主要内容:2.9.1 有机氟材料概述2.9.2有机氟的基础原料和基本单体制备2.9.3 氟树脂2.9.4 氟橡胶2.9.5 思考题2.9.1 有机氟材料概述氟是自然界最活泼的一种非金属元素,电负性最高,为3.9,能分解水,生成臭氧和HF,黑暗中就能与氢直接化合,几乎能直接与所有金属、非金属元素化合。
含氟的化学物质一般都具有特殊性质,例如热稳定性、化学稳定性、防水、防油等,因此引发了人们的研究兴趣。
我们常说的“有机氟”或“有机氟材料”指的是含氟的高分子。
含氟高分子的主链是碳链或碳杂链,氟原子作为碳原子上的取代基而存在。
碳原子上所有的氢全被氟替代,则成为只含有C和F原子的氟碳聚合物如聚四氟乙烯,又如四氟乙烯/六氟丙烯共聚物,可称之为全氟高分子。
部分氢原子被取代或含有其它原子(如氯),一般叫含氟高分子。
对于含氟高分子材料的分类较简单,可分为:含氟聚合物的单体,都是乙烯基衍生物,合成单体的路线,通常都由HF加成或由HF制成氟氯碳烷(氟里昂、氟致冷剂)裂解而成相应的烯烃单体,聚合反应一般都是连锁聚合而获得含氟高分子。
2.9.2有机氟的基础原料和基本单体的制备一、无水HF合成合成含氟化合物的最基础原料是HF,自然界没有F2和HF天然存在,必须人工合成。
许多含氟精细化学品例如含氟的油、脂、表面活性剂,通常都要借助于F2合成,F2产生主要靠电解无水的HF获得。
所以,HF尤其是无水HF是含氟材料的最基本原料。
自然界氟的化合物主要是氟萤石(CaF2),中国占全世界已探明贮量的70%。
用于合成无水HF的CaF2要求越纯越好。
一般使用一级或特级的矿石精矿。
其生产过程是由萤石和浓硫酸高温反应获得,如下式所示,中间伴随着很多副反应。
CaF2 + H2SO4—> CaSO4 + 2HF四氟乙烯(TFE )是由二氟一氯甲烷裂解获得,因此其合成步骤分为二氟一氯甲烷的合成和二氟一氯甲烷的裂解两步: CHCl 3 + 2HF >CHClF 2 CHClF 2 >C 2F 4 + 2HCl三、三氟氯乙烯合成三氟氯乙烯(CTFE )是由三氟三氯乙烷脱氯制得,因此其合成步骤分为三氟三氯乙烷的合成和三氟三氯乙烷的脱氯两步: C 2Cl 6 + 3HF >C 2F 3Cl 3 + 3HCl C 2F 3Cl 3 >C 2F 3Cl + ZnCl 2四、全氟丙烯(六氟丙烯)合成全氟丙烯(HFP )一般采用C 2F 4或CHF 3裂解制备,通常采用C 2F 4裂解: 3 C 2F 4 >2C 3F 6SbCl 5Ni700-900o C-Cl 2 ZnSbCl 5Ni700-900o C偏氟乙烯(CH2=CF2 ;VDF)由乙炔和无水HF加成制得CHF2CH3,然后在氯气存在下热裂解而得:C2H2 + 2HF > CHF2-CH3CHF2-CH3>CH2=CF2 + 2HCl六、其他含氟单体除了上述主要的含氟单体,常见的还有氟乙烯(CHF=CH2)、全氟辛酸等。

氟元素的化合物概述氟元素是元素周期表第9位的元素,原子序数为9,符号为F。
氟元素在自然界中一般以氟化物的形态存在。
在化学中,氟元素可以与各种其他元素形成各种化合物。
本文将详细探讨氟元素的化合物。
氟元素与金属的化合物氟化钠(NaF)•化学式:NaF•特性:无色结晶固体,易溶于水,具有较高的熔点和沸点•应用:用于制备氟化铝、氟化镁等化合物;用于制取氟气;用作防蛀剂、杀菌剂•制备方法:可通过氟化氢与氢氧化钠反应制备氟化铝(AlF3)•化学式:AlF3•特性:白色结晶固体,无臭,难溶于水•应用:用作铝电解制取的助熔剂;用于陶瓷、玻璃工业;用作催化剂等•制备方法:可通过氢氟酸与氢氧化铝反应制备氟化镁(MgF2)•化学式:MgF2•特性:白色结晶固体,无味,几乎不溶于水•应用:用作光学材料、电介质材料;用于制备氟化铯、氟化镉等化合物•制备方法:可通过氢氟酸与氢氧化镁反应制备氟元素与非金属的化合物氟化氢(HF)•化学式:HF•特性:无色气体,有刺激气味,易溶于水•应用:用作制取氟化物;用于皮革和纺织品的酸性洗涤剂;用作硅酸盐玻璃的蚀刻剂•制备方法:可通过氢气和氟气在适当条件下直接反应制备三氟化砷(AsF3)•化学式:AsF3•特性:无色液体,有刺激性气味,能迅速水解•应用:用作电池的有机电解质;用于有机合成反应的催化剂;用于制备其他氟化物•制备方法:可通过氟气与三氯化砷反应制备六氟化硫(SF6)•化学式:SF6•特性:无色气体,无臭,不易燃烧,在大气中具有良好的稳定性•应用:用作高压电器的绝缘介质;用于电容器、线缆等电力设备•制备方法:可通过硫与氟气在高温下反应制备四氟化碳(CF4)•化学式:CF4•特性:无色气体,无臭,稳定性较好,不易燃烧•应用:用作等离子体刻蚀(干法蚀刻)的电子工艺气体;用作电容器、绝缘材料的填充气体•制备方法:可通过四氯化碳与氢氟酸反应制备氟元素与有机物的化合物氟氯烃(Freon)•化学式:CClF3、CCl2F2等•特性:氟氯烃是一类含氟有机化合物,有多种不同结构的氟氯烃。

有机氟材料的结构及其应用学生姓名:任丽丽指导老师:刘耀华(太原师范学院化学系092班太原山西)摘要: 高性能、低(无)污染是当今发展的主要趋势,氟树脂独特的结构特点使它具有很高的耐热性、耐化学性和耐候性,独特的电学性能,优良的表面性能和光学特性,从而使其成为可能同时具有这两项要求的材料之一。
本文主要阐述了有机氟材料的结构及其在各方面的应用,尤其是在涂料和皮革工业上的应用。
指出今后皮革化学品将会向着多功能、高质量、环保型的方向发展。
另外还对国内外有机氟材料的发展做了简单的一些介绍。
关键词:氟材料结构与性能涂料皮革工业氟树脂前言:近年来,有机氟材料已经被应用于很多行业,例如涂料、皮革工业、保护文物的行业等等。
有机氟聚合物优异的耐候性、耐腐蚀性、耐玷污性、耐化学品性、斥水斥油性、绝缘性等,被广泛地应用于文物保护中。
氟系涂覆材料,由于其优异的耐侯性、耐腐蚀性、耐热性、耐化学品性、防污性、斥水斥油性及低摩擦性等优良特性,成为化工设备、海上平台、大型船舶防护等极端恶劣环境中使用的最高技术涂料。
本文将对有机氟材料的结构和应用进行介绍。
1.氟化学简介及有机氟材料的结构特点1.1氟化学概述1.1.1引言含氟化合物是当前增长最为迅速的精细化学品之一,广泛应用在材料、农药、医药等领域,具有广阔的发展前途和强大的生命力。
氟元素被引入分子后,分子的化学性能会产生深刻的变化。
由于自然界中几乎不存在有机氟化物,因此这完全是一门地地道道的人工合成的化学新领域,从而给有机化学家提供了无限机会。
1896年氟代乙酸乙酯的合成标志着有机氟化学的开始,至今已有一个多世纪的时间。
在此期间,几次历史性的突破极大地促进了有机氟化学的发展,如本世纪三十年代氟利昂应用于制冷工业,二战期间曼哈顿工程的实施,五十年代高生理活性氟脲嘧啶的合成等等【l】。
氟元素具有高负电性,它形成的有机氟聚合物具有卓越的耐化学性、热稳定性,优良的介电、耐热、耐药品、不燃、不粘及摩擦系数小等性能,是综合性能极佳的合成材料【2】。

碳酰氟和氟化氢全文共四篇示例,供读者参考第一篇示例:碳酰氟和氟化氢是两种重要的氟化学品,它们在有机合成、医药、材料科学等领域拥有广泛的应用。
本文将对碳酰氟和氟化氢的物理性质、化学性质、用途及安全性等方面进行详细的介绍,希望能为大家对这两种化学品有更深入的了解。
碳酰氟(Carbonyl fluoride,化学式为COF₂)是一种无色、具有刺激性气味的液体,密度为1.876 g/cm³,沸点为8.77℃。
碳酰氟在常温下易挥发,极易与水反应生成碳酸氟化氢并释放氟化氢气体。
碳酰氟是一种有机合成中的重要试剂,它可以用于合成酸酐、酰胺、酯等化合物。
碳酰氟还可用于金属表面处理、医药中间体的制备等领域。
碳酰氟和氟化氢都是有毒化学品,对人体有一定的危害。
碳酰氟的蒸气可引起呼吸道刺激、头晕、咳嗽等症状,长期接触可引起中毒。
而氟化氢对眼睛、皮肤和呼吸道有强烈的刺激性,接触过量可导致化学灼伤。
在使用这两种化学品时,必须严格按照操作规程进行,并保证充分的通风条件。
第二篇示例:要的应用价值。
本文将详细介绍碳酰氟和氟化氢的性质、制备方法以及应用领域。
碳酰氟(Carbonyl fluoride)是一种无色气体,化学式为COF2,分子量为66.01g/mol,其结构中包含一个碳原子和两个氟原子。
碳酰氟在室温下为液态,熔点为-124°C,沸点为-85°C。
碳酰氟是一种有机氟化合物,具有刺激性气味,易燃易爆,对皮肤、眼睛和呼吸道有刺激作用。
碳酰氟主要用于有机合成中的氟化反应,例如可以将碱性氨基团转化为氟取代基团。
碳酰氟还可以作为氟代试剂,用于氟取代反应中的催化剂。
碳酰氟和氟化氢的制备方法主要有以下几种:1.碳酰氟的制备方法:(1)从氟甲酸铅盐和五氧化二磷反应制得。
(2)从六氟丙酸和亚磷酸酐反应制得。
(3)从氟和二氧化碳气体在高温下反应制得。
碳酰氟和氟化氢在化学领域具有广泛的应用价值。
碳酰氟可以用于有机合成中的氟化反应,可以将化合物中的碱性氨基团转化为氟取代基团,从而改变分子的性质和活性。

含氟(⼀氟,⼆氟,三氟)化合物合成总结和应⽤⾃从1956年第⼀次出现含三氟甲基的精神类⽤药氟⾮那嗪(Fluphenazine)和1957年⾸次引⼊第⼀个含氟抗癌药物5-氟尿嘧啶(5-Fluorouracil)以来[1], 半个多世纪过去了. 近⼗年来, 隨着氟化学研究的进展和对氟原⼦及含氟取代基的深⼊了解, 药物科学家正在进⼀步开掘含氟药物这座新药研发中的⾦⼭银矿(Scheme 1)[2]. 最新统计表明, ⽬前全球含氟药物年销售额在400亿美元左右, 全球销售前200名的药物中, 含氟药物就占了29个, 销售额总计320亿美元. 由此看来含氟药物的应⽤及研发前景相当可观, 约有25%~30%的新药研发是建⽴在氟化学原料产品基础之上的. 依⽂献报道, 约15%~20%的新药都含有氟原⼦或三氟甲基等基团(图1)[3]. 据我们统计,截⽌2013年底, ⼀共有163个含氟药物接受被美国⾷品和药物管理局(FDA)的批准上市[4], 这充分说明氟原⼦是除氯原⼦之外, 第⼆个最令药物化学⼯作者喜欢的卤素原⼦. 在药物研究中, 充分利⽤构效关系(SAR)的⽅法学探索和越来越多含氟中间体的可得性, 都为含氟药物的研究提供了巨⼤的推动. ⽏容置疑, 最近⼏年的有机氟化学的研究热潮必将为新⼀波的新药探寻提供更多的⽅法和⼿段, ⽽在氟化学领域有世界⼀流的中国研究团队的参与[5], 也必将会浓墨重彩地书写药物研发的崭新⼀页. 本⽂将近年来这⼀领域的研究做⼀简述概括, 希望能为现代药物的合成研发提供帮助[6].不同于其它的卤素, 氟原⼦由于其独特的电⼦结构, 它具有最强的电负性和与氢原⼦⼀般⼤⼩的原⼦半径, 因⽽也能更加⽅便合理地取代氢原⼦⽽进⾏药物分⼦结构的微调和修饰, 阻断易代谢位点从⽽改变药物代谢的途径及代谢速度; 并通过分⼦间氢键的作⽤, 延长药物在体内的作⽤时间, 提⾼药物的⽣物利⽤度和选择性. 最值得⼀提的是, 三氟甲基由于具有很强的吸电⼦性、亲脂性和稳定性等特点, 具有很强的疏⽔性⽽表现出理想的脂溶性, 具有更好的⽣物通透性和靶向选择性, 因此在许多上巿药物和临床药物常常含有三氟甲基的芳环或杂环分⼦[7].2011年美国FDA共批准了35个⼩分⼦化学药物, 其中有7个是含氟新分⼦实体(Scheme 2);2012年共批准了33个⼩分⼦化学药物, 其中有6个为含氟有机分⼦(Scheme 3); 2013 年⼀共有8个含氟药物获批(Scheme 4). 据不完全统计, 总共有数⼗个含氟药物进⼊了临床研究, 其中⼀些代表性含氟药物见Scheme 5, 如Merck临床三期的胆固醇转运蛋⽩(CEPT)抑制剂Anacetrapib和Lilly的Evacetrapib, 这两个临床药物都含有三氟甲基和氟的芳⾹烃结构单元, 预期是药物中的重磅炸弹(Blockbuster drugs). 另外Daiichi研发的⾼效鲨烯合成酶抑制剂DF-461据称效果⽐上市的HMG- CoA还原酶抑制剂Atorvastatin(Lipitor)还要好. 可见含氟药物的研究正处于⼀个相当令⼈⿎舞的发展局⾯[8], ⽽有机氟化学的突破为这⼀药物研发的加速提供了很好的帮助.从近三年获批的含氟药物结构来看, ⼀共有17 个是氟代芳⾹烃, 有6个是含有三氟甲基的芳⾹环. 其中有2个药物既有三氟甲基⼜有氟原⼦. 显然, 有机芳⾹烃的氟化反应对新药研发是极其重要的, 为此, 本⽂就近⼏年来芳(杂)环氟化及N (n=1, 2, 3)氟甲基化的研究进展和亮点并结合实例进⾏分析总结.芳烃和杂芳烃的氟化反应可分为亲电取代(正离⼦F+试剂)和亲核取代(负离⼦F-试剂)两类型. 氟化反应少不了使⽤氟化试剂, ⽽且⼀般都使⽤过量的. 许多氟化试剂, 包括能提供氟正离⼦和氟负离⼦的氟化物见Scheme 6, 除了常⽤的KF, ⼀般氟化试剂都不便宜, 尤其是⼀些制备困难的有机氟试剂, 价格尤为昂贵.1.1 利⽤含氮(胺基、酰胺等)芳(杂)环为原料使⽤含氮杂环的联苯衍⽣物, Sanford⼩组[9]在2006年⾸先发展了有效的氟化反应(Eq. 1), 在10 mol% Pd(OAc)2 存在的条件下, 选择性地引⼊了氟原⼦. 这也是在钯(II)催化下, 温和氧化条件下利⽤亲电的氟化试剂(F+, 不同于使⽤亲核氟试剂F-)成功进⾏的经由直接C—H活化、氧化氟化(Oxidative fluorination)的创新反应.2009年, Scripps的Yu等[10]利⽤钯盐催化的、邻位导向基诱导的C—H活化策略, ⽤F+试剂在NMP为促进剂的情况下有效地制备了邻位含氟的芳⾹化合物(Eq. 2).不难看出, 上述⼏个反应都是在邻位导向基(direc- ting groups)存在下钯(II)催化的亲电氟化反应, 反应经历了环钯化(Cyclopalladation)和亲电氟化两过程. 巧妙的是, 分⼦内氮原⼦从不同的形式(如吡啶中的N; 取代的胺和酰胺)作为供电⼦体, 参与了分⼦内环钯化过程, 其特点是形成有利的五元钯(II)络合物中间态(Scheme 8).在没有导向基存在的条件下, 钯催化的亲电氟化反应的机理如图所⽰(Scheme 9). ⾸先是通过转移⾦属化⽽形成的碳钯键, 由于不涉及到环钯化(cyclopallada- tion), 也不需要导向基, 因此反应底物的多样性就更加⼴泛, 缺点是底物要进⾏预官能团化, 必须引⼊适当的基团进⾏转移⾦属化(transmetalation). 随后通过亲电氟化试剂氧化将Pd(II)络合物转化为Pd(IV)的络合物, 进⽽通过还原消除⽣成C—F键.1.2 利⽤芳基锡烷/有机硼化物为原料利⽤铜盐催化, Sanford⼩组[13]发展了温和条件下的芳基锡烷和三氟硼酸钾的氟化反应(Scheme 11).最近, Hartwig ⼩组报道[14]了铜盐催化下, 利⽤F+试剂对有机硼酸酯的氟化反应(Scheme 12).反应条件相对温和, 虽然铜试剂和银试剂的⽤量较⼤. 与此同时, 通过对19F NMR的考查, 确证了Cu(III)氟络合物的存在. 鉴于有机硼酸酯易得, 这⼀新⽅法为制备氟代芳烃⼜提供了⼀个新⼿段.1.3 利⽤芳基锡烷为原料使⽤不同的银盐(AgOTf, Ag2O)催化剂, Ritter 等[19,20]在2009年和2010年先后⾸先发展了温和条件下的芳⾹锡烷的氟化反应(Scheme 14), 并取得了良好的实验结果.1.4 利⽤芳⾹碘化物为原料和卤素交换反应(Halex processes)Hartwig⼩组[21]发现在铜盐和AgF的作⽤下, 取代碘苯能够有效地进⾏卤素交换⽽制备相应的氟苯(Scheme 15). 据机理研究表明, 该反应有Cu(0)和CuF2的形成. 反应没有完成催化循环.1.5 利⽤取代的苯酚和苯酚衍⽣物为原料从取代的苯酚和含羟基的取代的杂芳烃为起始原料, Ritter ⼩组[25]利⽤新颖的脱氧氟化试剂, 成功进⾏了操作简易可⾏的酚类化合物的氟化(Eq. 5). 该转化具有反应的产率⾼、基团兼容性好等优点.2009年Buchwald教授[27]⾸先报道了以三氟甲磺酸酯为原料的钯催化的亲核氟化反应, 成功的关键是⾤⽤了空间位阻⼤的特殊膦配体, 并通过形成刚性T形的的单核三配位的钯(II)络合物⽽完成反应. 有趣的是, 在⼀些反应中发现有区域异构体产⽣, 产物以间位产物为主(Scheme 19). 虽然⽬前对形成区域异构体的机理并不⼗分清楚, 但通过氟负离⼦进攻在原位的(in situ)产⽣的苯炔活泼中间体的解释基本排除在外.1.6 利⽤芳⾹胺和酰替苯胺为原料芳⾹胺重氮盐的HF/吡啶(Sandmeyer reaction)处理和氟硼酸处理(Balz-Schiemann reaction)是制备氟代芳烃的⽼⽅法. 使⽤有机⾼价碘, Li 和Meng等[30]成功进⾏了免除⾦属的直接区域选择性氟化酰替苯胺衍⽣物, 将氟原⼦引⾄酰替苯胺基的对位(Scheme 21). 该⽅法具有操作安全、试剂易得、条件温和、产率较⾼等特点.1.7 利⽤芳基碘鎓盐和季铵盐为原料最近, Sanford ⼩组[31]也成功研究出相转移条件下, 铜盐催化的不对称⼆芳基碘鎓盐的亲核氟化反应(Eq. 8). 该反应具有条件温和、反应快速、产率⾼、选择性好、显⽰很好的基团兼容性以及没有位置异构体等优点.1.8 后氟化(Late-stage fluorination)反应制备药物和天然产物的氟代衍⽣物Ritter⼩组[19,20]利⽤他们率先发现的银盐催化的后氟化(Late-stage fluorination)反应策略, 成功制备了数⼗个重要的上市药物和天然产物的氟代衍⽣物, 其中包括氟代Taxol、氟代DOPA、氟代Rifamycin S和氟代Camptothecin等(Scheme 22).胡⾦波等[33]于2009年已将选择性⼆氟甲基化和单氟甲基化做了很好的综述. 相对于芳烃的三氟甲基化和芳⾹烃的氟化反应, 芳烃和杂芳烃的⼆氟甲基化研究略显薄弱, ⽅法学⽅⾯的研究也显零散. 但这⼀状况最近得到了很⼤的改观. 在这⾥我们对近年的新进展做⼀概括.虽然迄今为⽌, 仍然没有上巿的药物中含有⼆氟甲基(CHF2)芳杂环亚结构单元的APl, 但是⼆氟甲基独特的结构特点仍然吸引了许多药物化学家的注意. 在等排物为基础的药物设计中, CHF2不失为优秀的亲脂性的氢键供应者, 为传统的氢键供应源提供了新的选择, 并且同时能有效地改善药物的膜渗透性, 促进药物的吸收. 例如在抗丙肝病毒(HCV NS3)蛋⽩酶抑制剂的研发过程中[34], 成功地利⽤⼆氟甲基取代并模仿母体化合物中硫醇的功能. 在COX-2和5-LOX双重抑制剂的研发过程中[35], ⼆氟甲基可做为异羟肟酸中羟基的等排物.传统的⼆氟甲基(CF2H)的引⼊⼀般都是由芳⾹醛和DAST试剂的反应⽽完成的. 考虑到有机磺酸盐中C—S键(272 kJ/mol)⽐有机硼酸C—B (377 kJ/mol)键和有机分⼦中的C—C 348 kJ/mol)要弱许多, Scripps 的Baran⼩组[36]成功制备了含⼆氟甲基结构的亚磺酸锌盐(DFMS), 利⽤过氧化叔丁醇为引发剂, 有效地均裂C—S键, 成功地制备了CF2H游离基, 并成功开发了相关反应(Scheme 23).值得注意的是, Baran ⼩组对⼀些天然产物和上市的重磅炸弹药物, 如辉瑞制药的Chantix进⾏了后三氟甲基化(Late-stage trifluoromethylation)和后⼆氟甲基化(Late-stage difluoromethylation)过程, 在不同的反应条件下, 经过不同的反应机理, 在母体化合物上有效地引进了CF3和CF2H官能团(Scheme 24), 这不失为药物创新改造和开拓构效关系(SAR)研究的新⽅法和快速通道. 从某种意义来说, 后N (n=2, 3)氟甲基化也是将芳(杂)环电⼦云密度⼤或着是电⼦云密度⼩的C—H键(即反应的活性位点)通过亲电取代和亲核取代这两种截然不同的反应被C—CF3(或C—CHF2)健取代, 应该是阻⽌药物氧化代谢的极好⼿段, 也是提⾼药物⽣物利⽤度的⽅便策略.将著名的Ruppert-Prakash试剂(TMSCF3)经过硼氢化钠还原, Hartwig ⼩组[40]以70%的收率制备了新的⼆氟甲基化试剂TMSCF2H, 并成功应⽤于铜盐催化的⼆氟甲基化碘苯(Eq. 12), 反应的收率⾼, 操作简单易⾏, 对各种官能团具有良好的兼容性.类似于三氟甲基(CF3)、单氟甲基(CH2F)可以被认为是甲基的有⽤的⽣物电⼦等排体. 其原因是氟的强吸电⼦效应使得氟原⼦能避免甲基的代谢性氧化. ⽽且单氟甲基也被认为是羟甲基(CH2OH)和甲氧基甲基(CH2OCH3)的⽣物电⼦等排体. 据调查, 真正意义上的单氟甲基化⽅法⼏乎不存在, 尤其是通过产⽣单氟甲基游离基. 应该承认单氟甲基化是难度很⼤、富有挑战的研究热点, 但这⽅⾯的⼯作最近取得了很⼤的进展, 其中包括Scripps的Baran教授的新试(CH2FSO2)2Zn及⽅法[37](Scheme 26).使⽤活性的芳⾹酮如9-芴酮作为可见光下苄基氢的捕⾷物, Chen⼩组[38]⾸次报告了选择性产⽣苄基游离基并通过加成氟游离基⽽完成苄基氟化的催化循环(Eq. 14). 值得⼀提的是通过巧⽤、活⽤不同的芳⾹酮作为光催化剂, 有效⽅便地制备了多达数⼗个结构各异、官能团兼容的带有氟甲基⽀键的芳⾹烃. 应该指出的是这也是迄今为⽌苄基氟化反应的最新进展.传承优秀的中国氟化学研究团队数⼗年来科研成果突出, 硕果累累, 亮点不断, ⼀直深受国际学术界的赞叹, 最近中国科学院上海有机化学所的卿凤翎教授[5]已经将近年来(2009~2011年)三氟甲基化的研究进展做了较为深⼊的总结和归纳. 北京⼤学王剑波教授[42]最近也对经由三氟甲基⾃由基进⾏的三氟甲基化的⼀些基本理论问题做了很好的探讨, ⽽西班⽛的氟化学专家Grushin也对⾦属参与的芳烃三氟甲基化在Chem. Rev上发表了系统全⾯的综述[43]. 考虑到对三氟甲基化反应研究⽅兴未艾[44], 这⽅⾯的⾼⽔平研究论⽂层出不穷, 我们仅将近年以来三氟甲基化研究的亮点从简易⽅便即实⽤性, ⾼效催化即绿⾊性和安全放⼤即⼯艺合理性等⾓度做⼀点评, 挂⼀漏万有偏颇之处, 在所难免.近年来, 不少新型的三氟甲基化试剂, 如正离⼦型的、负离⼦型(Scheme 27)的和游离基型的CF3来源, 它们为进⾏亲电、亲核和游离基三氟甲基化提供了选择的可能, 并成为反应条件优化必须考虑的主要因素.过渡⾦属催化的三氟甲基化反应在有机氟化学家和⾦属有机化学家的联⼿努⼒下, 已经取得了突破性的进展(Eq. 15). 但不可否认, 和成熟的钯催化的交叉偶联反应(Suzuki, Heck, Negishi)相⽐较, 改善的空间仍然是巨⼤的, 其主要缺陷是⼀般情况下钯催化剂的⽤量较⾼(10%), 三氟甲基化试剂⼀般较为昂贵, ⽽且有些试剂为⽓体或低沸点⼩分⼦, 物理化学性质不理想也是⼤⽓臭氧层的破坏者, 再者, 过渡⾦属的含量在药物分⼦API中的控制也是必须考虑的因素之⼀. 从这个⽅⾯来讲, Scripps 的Baran⼩组[45]在Langlosi⼩组[46]前期⼯作基础上将便宜易得的三氟亚磺酸钠作为三氟甲基源, 过氧叔丁醇作为氧化剂⽽进⾏的杂环的直接三氟甲基化. 由于反应不需要⾦属催化剂, 能在室温下进⾏, 反应条件温和, 溶剂为⽔/⼄腈(也不需额外处理), 对各种取代基及官能团不需保护和耐受性好, 是⼀种实⽤价值很强且易放⼤和⼯业化的好⽅法, 堪称⼀⼤突破. 当然对⾃由基参与的化学反应, 反应热的控制和安全性评价是必不可少的.有趣的是, 该三氟甲基化反应是通过⾃由基中间体完成的, 因此反应的区域选择性并不是⾼度专⼀的, 从某种程度来说, 它对现存药物的三氟甲基化⽽带来的新的药物分⼦的多样性确是⼀件好事, 因为芳烃或杂环分⼦有多于⼀个的活性反应点, ⽽便利地引⼊三氟甲基改造现有药物分⼦不失为⼀种旧药改造的捷径. Scheme 28 列出了⼀系列经过后三氟甲基化(Late-stage trifluoromethylation)的天然产物和重磅炸弹, 我们相信许多药物研发的科学家也会逐渐学习接纳这⼀新⼿段.Gooβen 研究⼩组[50]随后报道了铜盐催化下, 使⽤Ruppert试剂(TMSCF3)以芳⾹胺为原料的桑徳迈尔反应制备三氟甲基化芳烃(Scheme 31). 有所不同的是, 他们使⽤的催化剂是⼀价铜盐, 其中以硫氰化铜(CuSCN)催化活性最⾼. 反应温度为室温, 有很好的官能团兼容性, 产率在40%~98%之间.王剑波等[51]⾸先报道了通过三氟甲基银(AgCF3)和芳⾹胺参与的桑德迈尔反应来完成胺基⾄三氟甲基的官能团转化(Scheme 32). 值得⼀提的是, 反应需要在-78 ℃下完成, 否则收率较低.三氟甲基银是通过AgF和TMSCF3来制备的, 有趣的是: 使⽤CuCF3 为试剂, 在相似条件下, 产物的收率不佳(37%). 避免使⽤超低温反应条件(-78 ℃)也许是将来反应优化、⾛向适⽤性的发展⽅向之⼀.Baran研究⼩组[45]在利⽤便宜易得的三氟亚磺酸钠进⾏芳烃三氟甲基化⼯作的基础上, 最近⼜成功地从三氟亚磺酰氯和锌粉在⽔为溶剂中制备了物化性能更加优越的新试剂三氟亚磺酸锌, 并成功应⽤于芳⾹烃的三氟甲基化反应(Scheme 34). 利⽤该试剂, Baran研究⼩组对⼀些天然产物,药物中间体也进⾏了后三氟甲基化(Late stage trifluoromethylation), 并取得了理想的实验结果. 肖吉昌⼩组[55]也⾸次报道了铜促的采⽤三氟甲基锍盐的三氟甲基化碘代芳杂(稠)环的新⽅法(Eq. 22). 该反应具有官能团兼容性好、反应条件温和、产率⾼等优点, 对不同杂环系统的三氟甲基化均取得了很好的结果. 据称反应机理涉及到铜还原锍盐产⽣活泼中间体CuCF3的单电⼦转移过程.值得⼀提的是, 2010 年Yu等[59]利⽤⼀系列杂环如吡啶、嘧啶、咪唑和噻唑为邻位导向基, 在Pd(OAc)2的催化和TFA为促进剂的联合作⽤下, 成功地利⽤C—H活化⽅法进⾏了芳环的三氟甲基化, 取得了良好的实验结果(Eq. 26).20世纪80年代⾄90年代初, 陈庆云院⼠领导的研究团队[60]先后发现了数个三氟甲基化的试剂[5],其中1991年报道的1,1-⼆氟-1-氯代⼄酸甲酯(MCDFA)在KF和CuI存在下在DMF溶剂中能有效地进⾏相应酯的热分解(100~120 ℃), 通过消除CO2和CH3X, 完成相应的三氟甲基化反应, 反应具有条件相对温和、产率⾼、试剂便宜易得等优点, 该⽅法被称为陈试剂(陈⽅法, Eq. 27), 并⼴泛被国内外学术界和药业应⽤于含三氟甲基的化合物的合成.2013年Senanayake等[61]使⽤陈试剂, 在研发抗感染药物中, 通过条件优化并使⽤控制滴加的⽅法、溶剂筛选, 确定了最佳反应条件,有效控制了放⼤反应中泡沫CO2以及三个副产物的形成,成功制备了关键中间体(Eq. 28). 值得⼀提的是, 鉴于1,1-⼆氟-1-氯代⼄酸甲酯(MCDFA)在⼯业⽣产中⼤规模⽣产, 其成本要⽐使⽤Ruppert试剂降低85%.在⼯艺研发GSK3β抑制剂AZD8027的过程中, AstraZenca的Witt等[64]⾯临着同样如何有效和⽅便地引⼊氟原⼦和三氟甲基的问题. 很显然, 通过⽤Selectfluor引⼊氟原⼦后缩合形成氟代嘧啶杂环是极不可取的(Scheme 38). ⼀是氟化试剂昂贵, ⼆是氟化产率低(50%~55%), ⽽且有中间体不稳定等缺点.改进的合成⽅法⾸先合成了含氟嘧啶和含三氟甲基的咪唑⽚断, 然后通过杂环的Ziegler偶联关键反应等有效制备了AZD8926, 改进过的合成⽅法更趋合理(Scheme 39), 产率更⾼. 同时把钯催化的胺化反应提前, 避免了消除痕量钯⾦属所带来的困扰和API的损失.经过这些年的不懈努⼒, 有机氟化学在诸多⽅⾯得到了迅猛的发展, 世界⼀流的研究⼩组你追我赶, 创新⽴异, 成果频出. 应该承认, 有机⾦属化学的进展、催化反应的应⽤是今天氟化学进展的主推动⼒. 最近在有机化学的期刊上很难看到不含有氟化学的论⽂. 许多有关氟化学的专著也陆续出版[65], 综述⽂章和研究论⽂也不断更新[66].在这⾥, 我们将近年来芳⾹烃氟化反应和芳⾹烃杂环化合物N (n=1, 2, 3)氟甲基化反应的研究进展⽤图⽰的形式进⾏概括总结(Schemes 40, 41), 并概述了近年来含氟药物的研究成果. 值得特别强调的是第(III)种反应类型即新颖的氧化氟化反应(Oxidative fluorination)和氧化三氟甲基化反应(oxidative trifluoromethylation)是近⼏年来的研究热点课题之⼀. 我们相信最近的氟化学进展必将为药物研发⼯作者进⾏⼴泛的药物合成研究开辟了崭新的道路. 毫⽆疑问, 它必将拓展含氟药物建筑全新分⼦的想象空间, 提供更多重要的⽅法和途径.从发展趋势和⽅向来看, (1)理想的氟化反应应该是⽆⾦属(Metal-free)参与的或者是⾼效的过渡⾦属催化的过程, 应该⼤⼒开掘新颖⾼效的催化系统; (2)从经济、绿⾊环保等⽅⾯和为⼯业化放⼤⽣产考虑, Pd等催化剂的⽤量应该尽量控制在0.5~1 mol%之间(⽬前⼀般为5~10 mol%左右); (3)氟原⼦和N氟甲基的引⼊先后(现在⼀般的氟化物原分离纯化较难, 区域选择性不好⽽附带各种含氟杂质; (4)利⽤便宜易得的氟试剂, ⽬前许多含氟试剂较贵; (5)操作简单的⼯艺, 反应应该有不怕氧不怕⽔等优点, 安全且重复性好; (6)反应应该具有⾼产率, 尤其是含氟中间体和产物料都是市场上购⼊含氟⼩分⼦), 理想的状态应该是后加⼊F, ⽬前缺陷是: 收率⼀般并不太⾼, 产物纯品纯度不够, 如何进⾏分离纯化对⼯艺开发也是⼀个很⼤的挑战, 因为⼀般⽽⾔, 含氟化合物在多数有机溶剂中的溶解度⽐母体化合物更佳, 这使得利⽤重结晶、打浆等⼿段富积含氟化合物更趋不可能.应该指出的是, ⽬前在候选药物结构单元中什么位置引⼊氟原⼦和三氟甲基团, 仍带有相当的随意性, 仍然不能利⽤分⼦设计和计算化学的⽅法得到确切的判断. 也就是说含氟化合物的⽣理活性与分⼦结构的构效关系只能在⼀定程度上进⾏预测, 没有形成系统的理论体系. 显然这也是对药物科研⼯作者在含氟药物的合成与创新、开发⾼效低毒的新型药物提出的挑战.综上所述, 经过这⼏年有机氟化学家、有机⾦属化学家和药物化学⼯作者的共同努⼒, 在许多世界⼀流实验室参与和不⽢⼈后的研发热潮中, 有机氟化学的研究⾯貌发⽣了根本性的变化,极⼤地推动了含氟药物、多肽蛋⽩质化学和化学⽣物学的发展, 对材料科学和理论化学的发展也产⽣了很⼤的推动(Scheme 42). 毫不夸张地说, 这些年是有机氟化学发展的黄⾦时代, 它所创造的崭新氟化学反应⼿段和策略,为药物化学家开拓含氟药物这座宝藏提供了机遇和创造空间,也激发和丰富了有机化学家和药物科研⼈员的创造性和想象⼒,含氟药物仍是今后很长⼀段时间药物研发的⼀个热点课题, 这也是摆在中国有机化学家和药物科研⼈员⾯前的⼀⼤机遇和挑战.声明:。

全氟和多氟烷基化合物全氟和多氟烷基化合物是一类重要的有机物质,其在生产和生活中有广泛的应用。
这些化合物在化工、医药、制冷、灭火等领域发挥着重要的作用。
因此,掌握全氟和多氟烷基化合物的化学性质、合成方法以及应用是非常必要的。
一、全氟和多氟烷基化合物的定义和基本特性全氟和多氟烷基化合物是一类氟化学中的重要分支,它是一种含有氟原子的有机分子,在分子结构上,其所有的氢原子都被氟原子取代,因此,全氟和多氟烷基化合物具有高度的稳定性和惰性,且具有良好的化学惰性和热稳定性。
二、全氟和多氟烷基化合物的合成方法1、电化学法利用无水氟化氢或氟化铯、氟化银等氟离子源作为电解液,通过电解或电还原等方法,可以制备全氟和多氟烷基化合物。
2、烷化法烷基氟化剂作为反应物,与烷基溶剂进行反应,可以制备全氟和多氟烷基化合物。
这种方法可以根据需要选择不同的烷基氟化剂和溶剂,实现目标产物的选择性合成。
3、氟化法氟化物作为反应物,与有机化合物进行反应,可以合成全氟和多氟烷基化合物。
这种方法操作简单、反应温和,但是需要注意安全措施,因为氟化物对人体和环境有一定的危害。
三、全氟和多氟烷基化合物的应用1、化工行业全氟化合物在化工行业发挥着重要的作用,例如作为表面活性剂、润滑剂、脱模剂等。
2、医药行业全氟化合物在医药领域中被用作抗癫痫药物、麻醉剂等。
3、制冷行业全氟烷基化合物在制冷行业中广泛应用,可以用于制作各种冷冻设备。
4、灭火行业多氟烷基化合物具有优异的灭火性能,可以用于灭火器和灭火系统中。
总之,全氟和多氟烷基化合物是一类非常重要的有机物质,其具有广泛的应用前景。
在化学领域的发展中,对全氟和多氟烷基化合物的合成和应用的研究也将会不断深入,为人们的生产和生活带来更多的益处。

有机氟化合物的合成及应用研究有机氟化合物是一类具有重要应用价值的化合物,广泛用于医药、农药、材料科学和有机光电器件等领域。
随着有机化学的不断发展,有机氟化合物的合成方法也日益丰富和研究深入。
本文将探讨有机氟化合物的合成方法和应用研究。
首先,有机氟化合物的合成方法多种多样,其中最常用的方法是亲电氟化和亲核氟化。
亲电氟化是指通过亲电试剂与底物反应,将氟离子引入有机分子中。
这种方法常用于合成含氟有机化合物,如芳香氟化合物和氟代醇等。
亲核氟化是指通过亲核试剂与底物反应,引入氟离子。
这种方法常用于合成含氟氨基化合物和含氟碳酸酯等。
除了亲电氟化和亲核氟化,还有一些其他的合成方法,如芳烃和氟化剂反应、有机锂试剂和氟化试剂反应等。
有机氟化合物的合成方法不仅仅限于以上几种,根据具体的底物和要求,可以选择不同的反应路线。
例如,可以通过氟化巴铁和有机锂试剂反应,得到含氟有机铁配合物;可以通过氟烷和亲核试剂反应,得到含氟醇;还可以通过交叉偶联反应,将有机氟化合物与其他官能团连接在一起。
这些合成方法的发展,为有机氟化合物的合成提供了更多的选择和可能性。
除了合成方法的研究,有机氟化合物的应用也是一个重要研究方向。
有机氟化合物在医药领域的应用尤为广泛。
一些含氟药物被证明具有良好的活性和药代动力学性质,能够用于治疗癌症、糖尿病、心血管疾病等疾病。
例如,含氟醇类抗癌药物已经成为化疗的常用药物,其抗癌活性和生物利用度优于传统的抗癌药物。
此外,有机氟化合物还可以用于合成荧光探针、放射性示踪剂和核磁共振成像剂等,为生物医学研究提供了重要工具。
在农药领域,有机氟化合物也发挥着重要作用。
一些含氟农药被广泛应用于农作物保护,能够有效地控制害虫和病原菌的繁殖。
这些农药具有高效、低毒性和环境友好的特点,有助于提高农作物产量和质量。
此外,有机氟化合物在材料科学和有机光电器件领域也有广泛的应用。
由于氟原子的特殊性质,有机氟化合物可以提高材料的热稳定性、电子传输性能和光学性能。


探究分析有机氟化工新材料摘要:氟化工材料在当今有着相当广泛的应用范围,下到普通生产生活,上到国家高端科技,都可以看到有机氟化工材料的应用身影。
本文从我国的基本国情展开讨论,对我国有机氟化工材料的应用发展历程进行了分析,并讨论了当今应用热潮中的几类有机氟化工新材料,让人们对该项工业有了更深的了解,并为将来有机氟化工工艺的发展方向提供了一定的参考依据。
关键词:有机氟化工含氟工业新材料应用自上世纪以来,有机氟化工工艺就成为了全球各个国家重点研究与发展的项目,为推动国家科技与经济的发展做出了非常重要的贡献。
特别是对于我国来说,自改革开放之后,各项建设事业取得了飞跃式的发展,而在各类建设项目的实施过程中,无可避免地需要使用到不同类型的有机氟化工材料。
到了今天,各种有机氟化工新材料更是种类繁多,为人们的生产生活带来了极大的方便,更促进了国家高端科技产业的迅猛发展。
一、有机氟化工新材料在我国的发展应用有机氟化工工艺在我国的起源相对于国际上一些早期的工业国家来说要晚许多,因此在发展起点上相比于国际先进水平更低,发展的空间相对来说更大。
有机氟化工材料在我国得到十分广泛的应用主要在改革开放以后表现得十分明显,人们的生产生活以及许多高端科技产业逐渐离不开有机氟化工材料的支持。
(一)我国有机氟化工材料的应用现状在当今,有机氟化工材料的应用已经渗透到社会上的每一个阶层,不同的领域。
从国防军事到医药卫生,从家居生活到实验教学,都会使用到相应的有机氟化工材料。
比如说在室内装饰工程中所使用到的涂料,有很大一部分就属于有机氟化工材料。
并且在一些药品生产过程中,也会加入一定量的有机氟化工材料,从而对药液起到较好的过滤作用。
总之,从普通的生产生活角度来说,有机氟化工材料已经得到了普遍应用,并且随着科技的发展以及人们生活水平的提高,对这类材料的应用要求也在迅速提升,急需更多安全、方便、环保的有机氟化工新材料来满足人们的使用需求。
(二)我国有机氟化工材料的应用发展趋势近年来,我国的经济发展取得了举世瞩目的成就的同时,生态环境受到了较为严重的破坏。

有机氟材料的发展与应用届别 09届系别化学专业化学姓名郭萌萌学号 2009121140二〇一一年六月有机氟材料的发展与应用-----有机氟的发展史及研究成果学生姓名:郭萌萌指导教师:刘耀华摘要: 有机氟材料具有优异的耐高低温、耐热、耐化学品、绝缘、抗粘、低摩擦、不燃和自润滑等性能,由于这些材料具有与其它材料无法比拟的优良性能,使其应用已也从最初的军工领域逐渐扩大到民用、工业领域,成为国民经济中不可缺少的新型高分子台成材料。
我国的有机氟化学研究始于上世纪50年代后期,当时是为了满足国防建设的需求,经过50多年几代人的努力,如今我国已经能够生产许多含氟产品如氟塑料、氟橡胶、氟里昂、含氟表面活性剂、含氟油脂、含氟医药和农药、氟碳代血液等,形成了初具规模的氟化学工业基础。
本文主要介绍了我国有机氟材料的发展历程、研究现状以及在各领域的应用。
关键词: 有机氟化学有机氟材料发展成果应用有机氟材料其所以成为当前世界各国普遍重视的一类新材料,并未研究这类材料而形成的一门专门的科学----氟有机化学,是与它在当代科学技术进步和经济发展中所起的巨大作用密切相关的。
近年来,含氟功能材料和众多精细氟有机化学产品的出现,以及氟化学基础研究的进展,展示了含氟材料和氟有机化学更广阔的前景。
1.我国有机氟化学的发展1.1 任务带学科----有机氟化学的兴起1896年氟代乙酸乙酯的合成标志着有机氟化学的开始,至今已有整整一个世纪的时间,在此期间,几次历史性的突破极大地促进了有机氟化学的发展,如本世纪三十年代氟里昂在制冷工业上的应用,二战期间曼哈顿工程的实施以及50年代高生理活性氟脲嘧啶的合成等[1]。
我国氟资源丰富,已探明萤石的储量约占世界总储量的四分之一,但直到上世纪50年代,氟化学在中国还是一片空白,50年代末,由于国际形势的变化,我国开始自行开发原子能技术急需一批特殊的含氟材料,由此开始了有机氟化学在中国的研究。
1963年科学院决定将氟化学的工作集中到上海,集中力量形成特色,当时上海市调拨一个葡萄糖厂给有机所,经改造做为扩试和批量生产的基地,在这阶段的任务多数是仿制,成功后再批量生产。
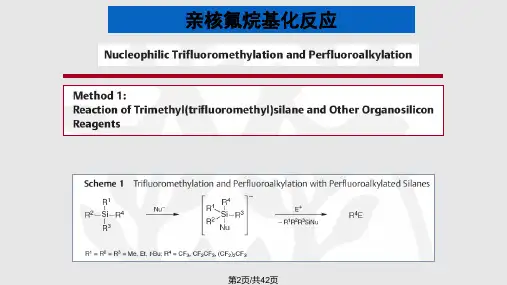
当代有机氟化学以下内容:来自于‹当代有机氟化学-合成反应应用实验›,自101页开始。
全氟烷基阴离子基本上可用于通常生成烷基或芳基阴离子一样的方法所产生,通过适当的C-H酸前体,用强碱脱质子或用还原性卤素(通常是溴、碘)金属交换,另外一种也是全氟世界所独有的方法即负离子或其他阴离子加成到全氟烯烃。
所有的全氟烷基阴离子由于受到氟取代的吸电子诱导效应(-I)而稳定,同时又受到氟原子的孤电子对对碳负离子中心的p-π电子排斥而去稳定。
对于β-氟碳负离子,负的超共轭效应可起到稳定化作用。
如果碳负离子并非处于自由的状态而是和金属(一个硬的路易斯酸),由于巨大的晶格能的释放趋向将强烈促使全氟烷基金属化物发生碎片化。
若存在β-氟原子,则将发生β-氟消除而产生末端全氟烯烃;若仅有α-氟原子,则发生α-氟消除而生成二氟卡宾,全氟芳基锂即使在低温条件下(一般-20*-40℃)也能发生消除,产生相应的芳基炔和氟化锂并伴随大量放热。
氟离子是很容易加成到全氟烯烃的,由于它将赴原子取代的SP3碳转化成SP2碳,而解除了p-π排斥引起的张力。
全氟丙烯或全氟烯烃的加成反应机理高度区域选择性的,他总是生成一个与带负电荷碳连有着最多碳原子数的阴离子。
氟离子很容易加成至全氟烯烃并生成一个碳负离子,用催化量的CsF处理全氟烯烃有时可以生成许多齐聚体的混合物。
五-三氟甲基环戊二烯阴离子生成的例子深刻反映了这种类型的反应。
它可以被应用于高度选择性的合成,例如五-三氟甲基环戊二烯基铯。
F 2C CHCF 3CF 3F 3C3CCF 33通过氟离子对全氟烯烃的加成产生全氟烷基阴离子的方法可以用于制备目的。
应用适当底物的脂肪族或芳环的亲核取代反应可选择性的引入全氟烷基。
对于芳香底物而言,离核的离去基团通常是氟离子,因此此类反应可改用催化量的氟离子。
催化剂或者是一个无机氟化物(CsF )或在一个电化学反应过程中由全氟烯烃的还原-脱氟产生。
长链全氟烷基锂化合物的生成通常是在更低的温度(<-78℃),他们通常是现场生成并立即和相应的底物(通常为羰基化合物如醛、酮或酯)直接进行反应。
若这些羰基化合物是手性的可以得到合理的对映选择性的过量产物。
NF F F FFCF 2=CFCF 3cat.TDAEN FF FF CF 3F CF 3但金属是软的路易斯酸,如锌、铜、镉,则她的全氟烷基金属化合物是稳定的。
由于金属-碳键更多的共价特征,一价铜金属烷基化物则很容易在较高的温度下被分离处理和进行反应。
三氟甲基锌稳定性较差,它可以用作亲核的CF 3的来源,既可以直接分离出来,也可以用锌在DMF 或THF 中与全氟烷基锂在超声波作用下现场生成。
金属烷基锌化合物可分别应用于Barbier 类型的反应,把催化交叉偶联反应及烯烃的全氟烷基氢化反应。
R F I +R Zn,CuI,THFR FR R F I+R F三氟甲基铜可用两种方法制备并进行反应:在150℃下一价铜盐与三氟乙酸盐反应或用铜粉与CF 3I 反应。
即使看起来稳定的CF 3Cu,也有证据表明存在CF 3I 和CF 2及CuF 之间的平衡,而这种平衡依赖于温度和溶剂。
这种平衡可以被用于分步地建立一个长链的全氟烷基铜配合物,它是通过一个:CF 2插入的机理,此反应可被添加少量的HMPA 而终止。
IO 2N CF 3Cu,DMF,HMPA0CF 3O 2NC 8F17BrO 2NDMF/NMP(1:1)800C CF 3O 2NCF 2CF 3O 2NNICl Me 3SiCF 3,CuI,KFDMF/NMP(1:1)250C 6h NCF 3Cl全氟烷基铜试剂最常用于与芳基溴或碘交叉偶合生成全氟烷基取代的芳环化合物.。
铜促进的三氟甲基化反应的一个缺点是生成全氟乙基衍生物,他在反应体系中及在纯化处理产物是很难被除去。
这个副产物的生成就是由于前面所提到的卡宾插入,对此我们可以利用降低反应温度或优化溶剂(如加入HMPA)来避免它的生成。
利用Me3SiCF3作为亲核三氟甲基的主要源泉的反应,可以在特别温和的反应条件下,即可现场生成CF3Cu.同样的方法(利用Me3SiC2F5)则可以成功地由芳基碘制备全氟乙烯基取代芳环的化合物。
铜促进的碘代芳烃和全氟烷基碘的交叉-偶合反应的机理类似于相应的卤代芳烃与有机亲核阴离子铜盐如(CuCN)之间的反应。
先生成一个溶剂的全氟烷基铜配合物(Ⅰ),随后与碘代芳烃配位并发生的配体交换。
该反应的成功大大依赖于溶剂对铜试剂的溶剂化能力。
DMF、吡啶、DMSO等溶剂可给出最高的产率。
该铜试剂对水解不敏感,反应中存在的有机还原剂。
反应对存在的羰基、氨基、羟基也不反应。
反应中被取代基团的活性依次是I>Br>ClR F-I Cu(溶剂L)+ICuL3RArRF+ICuL3不用金属的替代方法,用亲核还原活化全氟烷基碘,在低温时用有机还原试剂TDAE处理R f I所产生的R f-类物种(可能是一个电荷转移配合物R f I-TDAE)可被许多亲电试剂捕获,如与Me3SiCl 反应产生Ruppert试剂Me3SiCF3,或与羰基化合物反应生成醇。
从原子经济观点看,生成CF3-离子的最有效的方法使用强碱将价格低廉的CHF3去质子化反应。
但是CHF3沸点很低(-82.2℃),因此至少在实验室制备上必须处理气体;其次是为了防止CF3-碎片化,在他生成后要立即稳定或捕获它。
后来终于发现合适的溶剂与强碱结合的反应体系,如DMF 和KO tBu 、KN(SiMe 3)2以及DMSO/KH 等生成的CF 3-可与DMF 结合,他生成的半缩醛胺可被用于亲核的三氟甲基阴离子的储存库。
R F I +Me 2N Me 2NNMe 2NMe 2Me 3SiCl,二甘醇R F SiMe 3OCF 3F CF 3SO 2Cl C 4F 9iI,TDAE,甘醇二甲醚,-300C,2hS C 4F 9O O OCF 3R FI,TDAE,甘醇二甲醚,-300COHCF 3R F最近这一方法(CHF 3/DMF/强碱)又得到了进一步的拓展。
用全氟缩酰胺(可方便地用于吗啡啉或N-苯基哌嗪反应制备)作为三氟甲基化试剂,它是稳定的而且起始原料也不贵。
ONONHONOSiMe 3CF 32)0C,5hRSCF 3F 3COSiMe 3R 1R 2全氟烷基硅试剂:近几年来,Me 3SiCF 3及其全氟同系物Me 3SiR f 已经成为最常用的亲核全氟烷基化试剂。
被称为Ruppert 试剂的Me 3SiCF 3由Ruppert 合成于1984年,作为有活性的亲核三氟甲基化试剂则是由Prakash 及其同事们系统发展起来的。
它可用CF 3I 或CF 3Br 在各种还原剂如TADE 、P(NMe 2)3或Al 存在下,用CF 3Br 与Me 3SiCl 反应制备CF 3SiMe 3.CF 3Br+P(NMe 2)3+Me 3SiClCF 3SiMe 3OP(NMe 2)3CF 3Me 3SiCF 3KOBu t ,DMF Mg,Me 3SiCl DMFPhSK +3+PhS(O)CFPhSO 2CF +[O][O]CHO 1.Me 3SiCF 3,cat.Bu 4NF,THF,-200COHCF 3Bu tOOOBu t1.Me 3SiCF 3,cat.Bu 4NF,THF,-200CHO 3COHOMe 3SiCF 3,cat.40Me 3 1.Me 3SiCF 3,cat.Bu 4NF,THF,-200CF33OOATPH,Me 3SiCF 3,KOBu t庚烷/CH 2Cl 2,-78 toOOCF 3在氟离子催化下,如Bu tNF 或甚至是路易斯碱,Me 3SiCF 3可高产率地转化为CF 3-并与众多亲电底物(如羰基化合物)反应。
反应机理是生成一个类似于碳阳离子的烷基三甲基三氟甲基硅酯阴离子物种,它随后在一个自活化链反应过程中将CF 3-转移至羰基,这一反应可由少量的氟离子引发。
一些硅酯中间体物种已被成功的分离并用NMR 及X 晶体衍射证实。
它与许多其他一些有机硅试剂明显的不同是Me 3SiCF 3的加成反应不被许多路易斯酸催化引发。
三氟甲基对醛酮和其他一些羰基化合物的亲核加成反应首先生成相应的三甲基硅醚,随后被水解成相应的醇,反应条件温和,因此这一方法被广泛应用,对一些敏感底物同样也适用。
与其他一些方法相比,利用硅试剂进行氟离子引发的三氟甲基化反应对一些烯醇化物也能发生。
对一些α,β-不饱和底物,则优先发生羰基上的1,2-加成,若氟原子配位了一个较大的路易斯酸如ATPH ,则得到选择性的1,4-加成产物。
3这一试剂药物化学中最新的应用是青藁素化合物的三氟甲基化,三氟甲基的引入提高了它的药理性质。
30.2eq.Bu 4NF.3H 2O在较低的反应温度下利用手性氟离子源可以实现对前手性羰基化合物对映选择性的三氟甲基化。
但是,ee 值不高。
用非极性溶剂如甲苯、戊烷或二氯甲烷代替极性溶剂作为此类反应的介质,可以将各种各样的酯(芳酯、脂肪类酯、烯醇化的或非烯醇化的)全部转化成相应的三氟甲基酮。
OMe OMe 3SiCF 3,cat.Bu 4NF,甲苯;-780C to rt,18hCF 3O许多羰基化合物有足够的亲电活性可与Me 3SiCF 3进行反应,而许多的含氮亲电试剂必须用一种方法予以活化。
亚硝基苯这一简单的羰基化合物的杂原子类似物很容易与Me 3SiCF 3反应生成一个类似于羰基化合物的加成产物。
在同样条件下的亚胺却没有足够的活性,它们需要被如氮杂环丙烯化的立体张力或连有吸电子基的氮原子如硝酮或连在亲电的碳原子所活化。
非活化的亚胺在N-三甲基硅咪唑作为活化剂存在下,方可与Me 3SiCF 3反应,但是产率仅仅中等。
NO Me 3SiCF 3,cat.Bu 4NF,THF;0COSiMe 3CF31Me 3SiCF 3,Bu 4NF,THF N1F 33N ArCF 3F 3CMe CF 3CF 3F 3C NHAr利用对甲苯磺酰基可对亚胺进行活化,该基团在完成了三氟甲基化加成反应后可被除去,最后生成α-三氟甲基胺。
N-亚磺酰基也可顺利活化亚胺。
因此,借助于手性的N-亚酰基亚胺就可以完成高立体选择性的三氟甲基化,生成的亚磺酰基胺很容易被水解转化为带三氟甲基的手性胺,这些化合物在药物化学中是颇受重视的中间体。
NH SO 2Tol Me 3SiCF 3,Bu 4N+Ph 3SiF 2-,THF;0-50CN H CF 3SO 2TolBu t S N H O Me 3SiCF 3,Bu 4N +Ph 3SiF 2-,THF;-550C Bu t N HCF 3O 4M HCl,MeOH.rt +H 3N R F 3CCl - 运用类似的操作方法,Me 3SiCF 3也可用于许多硫亲电试剂的亲核三氟甲基化反应。