Weinreb酰胺的制备及应用研究进展
- 格式:docx
- 大小:368.38 KB
- 文档页数:9

有机合成中碳链上增加⼀个碳原⼦的⽅法⼀、以甲醛或甲醛等价物为底物进⾏反应增加碳链1、羟醛缩合反应(Aldol condensation)醛酮在碱性条件下得到烯醇盐和另⼀个羰基化合物缩合得到β-羟基醛酮的反应。
当利⽤甲醛作为底物时则底物增加⼀个碳。
Evans羟醛缩合反应,Abiko-Masamune羟醛缩合反应,Mukaiyama羟醛缩合反应2、Arens-van Dorp反应烷氧基⼄炔在强碱条件下对醛酮加成得到烷氧基炔甲醇的反应。
3、Stobbe condensation丁⼆酸⼆⼄酯及其衍⽣物和羰基化合物在碱性条件下进⾏缩合的反应。
4、Knoevenagel缩合反应羰基化合物和活泼亚甲基化合物在胺催化下缩合的反应。
5、Stetter反应醛和α,β-不饱和酮在噻唑盐的催化下反应制备1,4-⼆羰基化合物的反应。
噻唑盐是氰离⼦的安全替代试剂。
此反应也被称为 Michael-Stetter反应,机理和安息⾹缩合类似。
此反应直接利⽤甲醛作为底物的报道较少,但是有⽂献报道利⽤糖作为甲醛替代物进⾏反应可以得到多⼀个碳的1,4-⼆羰基化合物。
【J. Am. Chem. Soc. 2013, 135, 8113–8116】6、Barbier反应在有机⾦属试剂存在下,羰基化合物可以迅速与其反应,这类反应被称为Barbier反应。
7、Grignard反应(格⽒反应)格⽒反应有多多种⽅式增加碳链,可以考虑以甲醛为底物和格⽒试剂进⾏反应增加⼀个碳链得到醇,也可以以⼆氧化碳为底物进⾏加成得到羧酸,或者直接利⽤甲基格⽒试剂对其他亲电试剂进⾏延长碳链。
8、Kagan-Molander偶联反应9、贝蒂反应(Betti Reaction)酚与芳⾹醛和伯胺作⽤得到 α-氨基苯甲酚类。
这个反应可以视为苯酚的Mannich反应。
10、Mannich反应1903年,B. Tollens和von Marle发现苯⼄酮和甲醛,氯化铵反应可以⽣成三级胺。

Weinreb酰胺的合成方法进展易亮;齐永菲【摘要】Weinreb酰胺是合成一些结构有意义或有生物活性的手性化合物的重要中间体,它可与有机金属试剂反应生成酮,又可被二异丁基铝氢(DIBAL-H),氢化铝锂(LiAlH4)等还原成醛,现已被普遍应用于全合成化学、药物化学以及生物有机化学等领域。
因此,研究Weinreb酰胺的合成具有重大意义。
本文综述了以羧酸、羧酸衍生物等为原料制备Weinreb酰胺的方法并对制备Weinreb酰胺的方法进行了展望。
【期刊名称】《黑龙江科技信息》【年(卷),期】2015(000)013【总页数】2页(P37-37,38)【关键词】Weinreb酰胺;合成;研究进展【作者】易亮;齐永菲【作者单位】哈尔滨理工大学,黑龙江哈尔滨 150000;哈尔滨理工大学,黑龙江哈尔滨 150000【正文语种】中文酰基化试剂[N-甲基-N-甲氧基酰胺(Weinreb酰胺[1])]1是由Weinreb和Nahm 于1981年发现的一类重要的化学合成中间体。
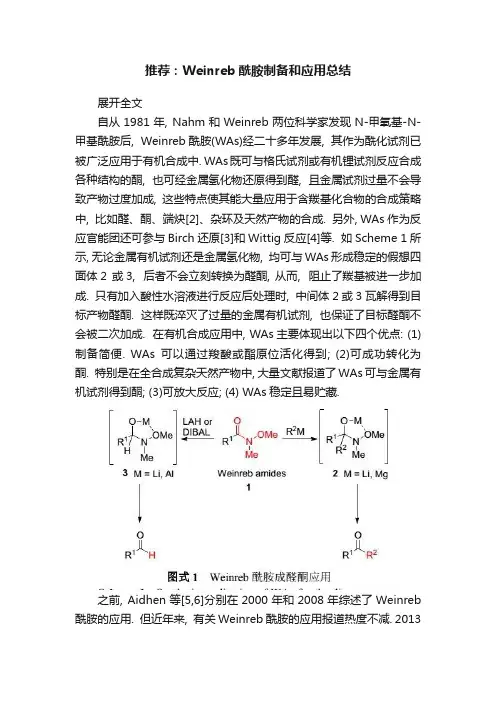
Weinreb酰胺类化合物作为重要的化工原料和药物中间体,其应用已遍及化工、医药、农药、天然产物、材料等方面的合成,这种酰胺的优势就在于其与过量的金属有机试剂反应,只生成酮,而不会继续生成醇,其反应过程见scheme1。
由scheme1可见在反应过程中生成了稳定的金属螯合物2,该螯合物不会离解产生羰基化合物,过量的金属有机试剂也不能使其进一步发生反应,只能通过酸解生成相对应的酮。
Weinreb酰胺同时也容易被金属氢化物还原生成醛,其反应过程见scheme2。
值得一提的是金属有机试剂或金属氢化物与具有光学活性的Weinreb酰胺反应时,Weinreb酰胺的构型可保持不变。
本文就近年来Weinreb酰胺的合成研究进展进行综述。
直接从羧酸出发与N-甲基-N-甲氧基胺一步合成Weinreb酰胺是较理想的制备方法,但是该反应中羧基酰胺化反应的活性弱,很难与N-甲基-N-甲氧基胺直接缩合,羧基要先活化后才能与N-甲基-N-甲氧基胺反应生成Weinreb酰胺。

金属催化条件下酰胺类化合物的合成研究中文摘要酰胺键在生命体构建过程中所扮演着极为重要的角色。
例如,蛋白质(包括酶)中的肽键就是酰胺键。
同时,酰胺键也广泛存在于很多天然产物和现代医药分子中。
因此,很多生物学家和化学家一直将酰胺键的构建放在首要位置。
尽管酰胺键的构建方法有很多种,但是其中的大部分还是涉及到偶联剂的使用,这就增加了合成的成本。
因此,两者之间的矛盾促使我们继续探究催化条件下合成酰胺键的方法。
本文以金属催化条件下,合成酰胺类化合物这个问题为中心,分别合成了α-苯磺酰胺基酮和含异噁唑啉环的酰胺。
与此同时,对以上两种产物的生成机理加以研究。
具体内容如下:一:异噁唑啉环在有机合成以及药物研究中意义重大,含异噁唑啉环的酰胺类化合物为医药研究提供了更多的选择。
在室温下,β,γ-不饱和酮肟在碘苯二乙酸(PIDA)和三氟甲烷磺酸锌(Zn(OTf)2)共同作用下,发生σC-C键裂解,得到氧化腈和互补的含烯键的碳正离子中间体。
后者与体系中的腈反应,得到N-烯丙基酰胺。
接下来二者发生分子间的1,3-偶极环加成反应,得到了含异噁唑啉环的酰胺。
二:鉴于氮自由基难以生成,且与炔键的加成比较困难,我们设想以N-氟代双苯磺酰亚胺(NFSI)为氮自由基前体,与芳基丙炔酸反应,能否合成双苯磺酰基取代的炔烃。
实验结果表明,生成了α-苯磺酰胺基酮。
反应的机理可能为:在一价铜盐和银盐及配体存在下,N-氟代双苯磺酰胺(NFSI)生成磺酰胺自由基,该自由基区域选择性地加到炔银中间体中距离芳环比较远的一端,得到高活性的烯基自由基。
该中间体紧接着发生1,4-芳基迁移伴并伴随脱二氧化硫。
然后,发生氧化/水的亲核进攻/半频哪醇重排,最终生成α-苯磺酰胺基酮。
关键词:金属,催化,含异噁唑啉环的酰胺,α-苯磺酰胺基酮Metal-Catalyzed Synthesis of AmidesAbstractUndoubtedly, amide bond is essential to sustain life. For Example, it is the most basic covalent bond in peptides(e.g. enzymes). Meanwhile, amide bond can be seen in a variety of natural products and modern pharmaceutical molecules of interest. So, numerous biologists and chemists have given their priority to the construction of amide bond. However, despite the abundance of the building methods, most of them involve the use of stoichiometric amount of coupling reagents, which increases the expense accordingly. Hence, this contradiction motivates us to continue the exploration of developing novel metal-catalyzed methods for achieving amide bonds. This paper mainly centers on the preparation of α-benzenesulfonamido ketones and amides containing isoxazoline. The details are as follows:1. Isoxazolines have been reported to be of synthetic and pharmaceutical importance, and amides containing isoxazolines provide more posibility for medical research. With the synergistic effect of (Diacetoxyiodo)benzene (PIDA) and Zinc trifluoromethanesulfonate, the σcarbon-carbon bond of β, γ-unsaturated ketoximes cleavaged to give intermediate nitrile oxides. The counterpart so-obtained reacts with nitriles, and elaborated N-allylamides. This is followed by intermolecular 1,3-dipolar cyclization of nitrile oxides and N-allylamides, infrequently affording the amides containing isoxazoline.2. In view of the fact that N-centered radical is difficultly available and is reluctant to react with carbon-carbon triple bond, we envisioned that maybe N-Fluorobenzenesulfonimide (NFSI) can be utilized as precusor of N-centered radical and the latter then reacted with arylpropiolic acid to deliver benzenesulfonimide-functionalized alkynes. Happily, we obatined α-benzenesulfonamido ketones. The poposed mechanism is as follows: NFSI is activated by cuprous salt, generating nitrogen-centered radical which regioselectively added to the triple bond of alkyne silver intermediate, distant from the aryl group. Subsequent 1, 4-aryl migration motivates the loss of sulfur dioxide. Then, a oxidation/nucliphilic attack of water/semi-pinacol rearrangement sequence finally furnishes α-benzenesulfonamido ketones.Keywords: metal-catalyzed,amides containing isoxazoline, α-benzenesulfonamido ketones目录中文摘要 (I)Abstract (II)第一章酰胺键的应用与合成概述 (1)1.1 前言 (1)1.2 传统合成酰胺键的方法 (2)1.2.1 偶联剂作用下羧酸和胺的反应 (2)1.2.2 Beckmann重排 (3)1.2.3 Ritter反应 (7)1.2.4 Ugi多组分反应 (9)1.2.5 腈的水解 (11)1.3 较新颖的合成酰胺的方法 (13)1.3.1有机化合物或过渡金属催化羧酸和胺直接生成酰胺 (13)1.3.2羧酸替代物和胺的反应 (16)1.3.3胺替代物和羧酸的反应 (24)1.3.4其他类型反应 (25)第二章由β,γ-不饱和酮肟合成含异噁唑啉环的酰胺 (28)2.1 异噁唑啉类化合物的用途 (28)2.2 异噁唑啉类化合物的合成方法 (28)2.3 工作背景及研究目的 (32)2.4 实验结果和机理研究 (33)2.5 实验部分 (40)2.5.1 实验仪器 (40)2.5.2 试剂和溶剂 (40)2.5.3 底物β,γ-不饱和酮肟的制备 (40)2.6 部分底物谱图和产物谱图数据 (41)2.6.1 1l的谱图数据 (41)2.6.2 产物的谱图数据 (42)第三章α-磺酰胺取代的酮的合成研究 (50)3.1 α-磺酰胺取代的酮的合成方法 (50)3.2 研究背景和研究目标 (54)3.3 实验结果和讨论 (55)3.4 反应机理研究 (58)3.4.1 自由基捕截实验 (58)3.4.2 对照实验 (58)3.4.3 反应机理总结 (58)3.5 实验部分 (59)3.5.1实验仪器 (59)3.5.2实验步骤 (59)3.6 产物谱图数据 (61)参考文献 (68)在学期间的研究成果 (80)致谢 (81)第一章酰胺键的应用与合成概述1.1 前言据报道,截至2006年,酰胺键在2/3的候选药物中被发现[1]。

学年论文题目: Weinreb酰胺的制备及应用研究进展学院:化学化工学院专业: 11级化学(师范)指导教师:学生姓名学号:2014年 5月 30日目录摘要------------------------------------------------------------------------------第3页关键字---------------------------------------------------------------------------第3页1.Weinreb酰胺简介------------------------------------------------------第3页2.Weinreb酰胺的合成方法:-------------------------------------------第4页2.1有机锌试剂制备weinreb酰胺的方法------------------------第4页2.2以羧酸为原料的合成法-----------------------------------------第5页2.3以羧酸酯为原料的合成法----------------------------------------第6页2.4以酰氯为原料的合成法-------------------------------------------第6页2.5以酰胺为原料的合成法-------------------------------------------第7页3. weinreb酰胺的应用进展----------------------------------------------第7页3.1用weinreb酰胺合成酮---------------------------------------------第7页3.2weinreb酰胺还原反应--------------------------------------------第7页3.3weinreb酰胺水解反应--------------------------------------------第8页4.总结:-------------------------------------------------------------------------第8页参考文献:---------------------------------------------------------------------第8页Weinreb酰胺的制备及应用研究进展摘要:Weinreb酰胺是一类十分重要的酰基化试剂,在许多天然产物的合成中有着的广泛的应用,其合成的方法有很多种,在此介绍羧酸、羧酸酯、酰氯、酰胺为原料和用有机锌试剂制备weinreb酰胺的方法。

翻译文献:One-Pot Synthesis of Pf-Disubstituted a,「-UnsaturatedCarbonylCompoundsMasaharu Sugiura,* Yasuhiko Ashikari, and Makoto NakajimaP位二取代的a, 0-不饱和羰基化物的一锅法合成摘要:TiCl4催化酮的羟醛缩合反应从而一锅合成P,P-二取代的a, P-不饱和携基化合物,其中酮为羟醛的受体,且在消除阶段会有钛氧基团从钛-羟醛配合物上离去。
在消除阶段使用添加剂(如二甲基甲酰胺、四甲基乙二胺和毗啶)是很重要的。
正文:p,P-二取代的a,P-不饱和携基化合物是本身就是有用的化合物,并且可作为在携基的P位上可以构建四取代或三取代的手性碳原子中心原料。
各种各样的天然产物可以通过具有这种官能团的化合物来合成。
我们也报道了路易斯碱催化的不对称共轭的P,P-二取代a,P-不饱和三氯烯酮。
酮的Horner-Wadsworth-Emmons组合反应和后续经Weinreb酰胺作用的转化为烯酮是广泛用于制备环P,P-二取代的链状烯酮。
酮的亚磺酰化/脱亚磺酰化,有机金属试剂经加成为P-氨基烯酮,P-烷硫基烯酮或P-烷氧基烯酮,有机金属试剂的共轭加成,三级炔丙醇或其乙酸酯,三烯丙基醇的氧化重排都被利用了。
最近,Donohoe和其同事报道了一个基于乙烯Weinreb酰胺的Mizoroki Heck 反应,该方法具有很有效的立体选择性。
然而,这些先例都需要经过几个反应步骤。
醇加成到的单酮上后脱水(羟醛缩合)可为合成P,P-二取代的a,P-不饱和携基化合物提供一种简单的方法,而且能较容易获得原料;然而,这反应还尚未进行了系统的研究,因为作为亲电试剂(醛醇受体)的酮的反应活性相对较低,反应可逆性强,而且容易生成不必要的自身羟醛缩合产物或交叉羟醛缩合产物。
在很少的例子中,Tanabe和他的同事报道的非常有用的非对映选择性TiCl4 / Bu3Nn催化两酮之间或苯基酯/苯基硫酯和酮之间的羟醛缩合反应。

weinreb酰胺的制备
Weinreb酰胺是一种重要的羰基合成试剂,可以用于合成酮和醛化合物。
下面是Weinreb酰胺的制备方法:
1. 材料:
- N,N-二甲基甲酰胺(DMF)
- 三氯甲烷(CHCl3)
- 碳酸二甲胺(DMA)
- 甲酸
- 二甲基氨基乙基氯化铜(CuCl2·2DMF)
2. 实验步骤:
a. 准备一个干净的干燥的反应瓶,用氮气干燥瓶内空气。
b. 向瓶中加入DMF(1.2当量),然后加入三氯甲烷(2.2当量)。
c. 向瓶中加入碳酸二甲胺(1当量)。
d. 将甲酸(1当量)添加到瓶中。
e. 加入二甲基氨基乙基氯化铜(0.1当量)。
f. 将反应瓶密封,并在室温下搅拌反应约24小时。
g. 完成反应后,用水稀释反应混合物,并用氯化钠饱和。
h. 用冷水洗涤有机相,然后使用氯化钠饱和水溶液洗涤,最后用氯化钠饱和溶液洗涤。
i. 使用旋转蒸发仪将有机溶剂蒸发掉,得到Weinreb酰胺产物。
j. 需要根据具体的实验需求进行进一步纯化和抽提等操作。
请注意,这只是Weinreb酰胺的一种制备方法,实际操作中可
能还会有其他变体的方法。
此外,在操作化学试剂时,请严格遵循操作规程和实验室安全指南。

非经典 Wittig 反应的最新进展荣红英;黄文华【摘要】综述了非经典 Wittig 反应的最新进展,包括酯、酰胺、酰亚胺、酸酐、唑酮等非醛酮类羰基化合物的 Wittig 反应。
通过改变磷叶立德的结构或设计成分子内 Wittig 反应,可合成各种杂环化合物和药物前体。
%Recent progress of non-classical Wittig reaction was reviewed in this paper,including Wittig reac-tion of some non-aldehyde and non-ketone carbonyl compounds such as ester,amide,imide,anhydride,and ox-azolone.Through modifying the structure of phosphorus ylide or designing an intramolecular reaction,a variety of heterocyclic compounds and drug precursors could be synthesized.【期刊名称】《化学与生物工程》【年(卷),期】2015(000)010【总页数】7页(P1-7)【关键词】Wittig反应;酯;酰胺;酰亚胺;酸酐;唑酮【作者】荣红英;黄文华【作者单位】天津大学化学系,天津 300072;天津大学化学系,天津 300072【正文语种】中文【中图分类】TQ203.9;O621.3Wittig反应[1]是有机合成中生成碳碳双键最常用和最可靠的反应之一,最经典的形式是磷叶立德与醛或酮反应生成烯烃(图1)。
磷叶立德又称Wittig试剂,分为稳定(R 1和R2至少有一个为吸电子基)、半稳定(R1或R2为芳基或烯基)和不稳定(R1和R2为烷基或氢)的磷叶立德,反应活性依次升高。

Weinreb酰胺与格氏试剂反应的机理如下:
1. 格氏试剂的形成:卤代烷(如卤代烃)与金属(如镁)在无水乙醚或四氢呋喃等有机溶剂中反应生成格氏试剂。
这个步骤通常需要在惰性气体(如氮气)下进行,以避免水和氧气的干扰。
2. 格氏试剂的亲核加成:生成的格氏试剂作为亲核试剂与酰胺中的酰基发生亲核加成反应。
格氏试剂中的金属负离子攻击酰基的羰基碳,形成一个暂时的中间体。
3. 中间体的形成:格氏试剂的亲核攻击导致一个负电荷在羰基碳上形成,同时酰胺中的氨基或胺基中的氢离子被金属负离子的氢替换。
这形成了一个中间体,其中羰基碳与金属负离子和新形成的氨基或胺基结合。
4. 中间体的消除:中间体中的负电荷通过质子化来中和。
这可以通过加入水或酸来实现,从而形成相应的酸性介质。
以上机理仅供参考,可以查阅相关文献资料获取更多信息。
推荐:Weinreb酰胺制备和应用总结展开全文自从1981年, Nahm和Weinreb两位科学家发现N-甲氧基-N-甲基酰胺后, Weinreb酰胺(WAs)经二十多年发展, 其作为酰化试剂已被广泛应用于有机合成中. WAs既可与格氏试剂或有机锂试剂反应合成各种结构的酮, 也可经金属氢化物还原得到醛, 且金属试剂过量不会导致产物过度加成, 这些特点使其能大量应用于含羰基化合物的合成策略中, 比如醛、酮、端炔[2]、杂环及天然产物的合成. 另外, WAs 作为反应官能团还可参与Birch还原[3]和Wittig反应[4]等. 如Scheme 1所示, 无论金属有机试剂还是金属氢化物, 均可与WAs形成稳定的假想四面体2 或3, 后者不会立刻转换为醛酮, 从而, 阻止了羰基被进一步加成. 只有加入酸性水溶液进行反应后处理时, 中间体2或3瓦解得到目标产物醛酮. 这样既淬灭了过量的金属有机试剂, 也保证了目标醛酮不会被二次加成. 在有机合成应用中, WAs主要体现出以下四个优点: (1)制备简便. WAs可以通过羧酸或酯原位活化得到; (2)可成功转化为酮. 特别是在全合成复杂天然产物中, 大量文献报道了WAs可与金属有机试剂得到酮; (3)可放大反应; (4) WAs稳定且易贮藏.之前, Aidhen等[5,6]分别在2000年和2008年综述了Weinreb 酰胺的应用. 但近年来, 有关Weinreb酰胺的应用报道热度不减. 2013年, Davies 等[7]还在利用N-酰基手性辅助基团不对称合成手性醛酮的综述文章中, 提到了开发手性WAs替代基团进行不对称催化工作. 鉴于Weinreb酰胺的诸多优点及其在合成上的广泛使用, 结合近年来的发展趋势, 本文综述了当前WAs的主要制备方法、最新应用进展及使用限制, 以期全面介绍WAs, 丰富有机化学家的合成手段.鉴于Weinreb酰胺在合成上频繁被使用, 其各种制备方法已被大量报道, Scheme 2所示. 一般, Weinreb酰胺可以从羧酸及其衍生物为原料, 比如酰氯、酯、内酯、酰亚胺和酸酐等, 与市售的N,O-二甲基羟基胺盐酸盐(DMHA)反应得到. 这其中, 羧酸与DMHA直接转化为WAs的制备策略, 操作最为方便而倍受关注, 因为这样可以避免先将酸转化为反应活性更高的羧酸衍生物(path a). 根据这个策略, 研究者们尝试了各种羧酸活化试剂, 比如: DCC, DEPC, HOBT, CBr4/PPh3, CDI, 烷基氯仿, BOP, EDCI, PPA, CDMT, HOTT, CPMA 及DMT-MM 等肽缩合试剂, 用于WAs的制备, 具体文献可见综述[6], 在此不做复述. 以上反应虽然可以有效制备WAs, 但有时也存在收率低, 反应时间长, 反应剧烈及分离纯化困难等缺点. 最近, 肽缩合剂T3P/DBU[8]被报道用于N-保护氨基酸(肽)WAs 的制备(Eq. 1). 由于, T3P具有低毒、反应温和、廉价及商品化等特点, 与碱DBU配合, 可对各种N-保护的手性氨基酸4, 甚至二肽,与DMHA缩合制备WAs衍生物5, 该反应收率高(>90%), 便于分离且不消旋.此外, COMU®作为第三代脲阳离子肽缩合剂也被成功用于N-保护氨基酸WAs的制备[9]. 不像HATU等苯并三唑类缩合剂, COMU®结构中不含三唑基团, 危险性极低; 在制备WAs反应中, 存在明显的颜色变化, 可裸眼判断反应进程; 反应副产物溶于水, 便于分离, 并且手性氨基酸的消旋化极低. 虽然有以上诸多优点, 但COMU®比较昂贵是一大缺点.在形成酰胺键的反应中, PPh3常与含卤化合物配合使用, 比如: NCS[10], NBS[11], Br2[12], BrCCl3[13], CCl4[14]等, 可以与羧酸反应有效形成酰胺键. 早先, PPh3/ CBr4组合已经用于WAs的制备[15]. 2010年, Kumar等[16]报道了利用PPh3/I2组合, 可活化羧基, 与DMHA缩合成WAs的反应(Scheme 3). 首先, 等物质的量比PPh3与I2 得到碘化鏻盐8, 后者与脱质子的羧酸形成酰鏻盐或酰碘中间体, 再与DMHA缩合制备9. 该反应在0 ℃进行, 便于操作, 收率在70%左右.2009年, Niu 等[17]报道了利用PCl3与DMHA反应得到P[NCH3(OCH3)]3(10), 后者可以在甲苯中直接与各种羧酸(芳香酸、脂肪酸及二元酸), 特别是位阻大的羧酸, 高收率制备WAs (Scheme 4).除了从羧酸直接活化制备WAs, 也可利用酰卤与DMHA缩合成WAs (path b). 可用于制备WAs的酰化试剂分别为SOCl2和Deoxo-Fluor, 它们可将羧酸先分别转化为酰氯和酰氟. 2013 年, Pace 等[18]报道了酰氯可与DMHA在生物溶剂2-MeTHF[19]和碱水组成的两相体系中制备WAs (Eq. 2). 由于2-MeTHF与水不互溶, 反应中生成的13 溶在有机相, 而盐酸以无机盐的形式与副产物溶于水相, 反应结束后只需简单分液、减压蒸出2-MeTHF即可得到纯净的WAs. 整个过程不需额外使用任何其他有机溶剂, 体现出很好的绿色化学特性.与酰氯相比, 酰氟的反应活性更像酯, 比酰氯要更加稳定, 因此反应条件不苛刻. 利用Deoxo-Fluor试剂将羧酸转化为酰氟后, 可用于制备WAs. Deoxo-Fluor试剂14已经用于WAs合成长链脂肪酮[20]. 最近发现, 14甚至可与血浆中的游离脂肪酸形成酰氟, 再与二甲胺反应得到类Weinreb酰胺. 该衍生化方法可用于GC-MS定量检测血浆中游离脂肪酸的含量[21]. 另外, Deoxo- Fluor试剂15也可用于合成4-氟吡咯烷WAs衍生物[22].2014年, Gupta 等[23]报道了从醇或醛合成三氯甲基甲醇16, 后者可经同系化-胺化反应制备多一个碳的WAs 17(path c), 收率达到75%~89% (Eq. 3). 该反应的底物适用性不是特别理想, 当R为除芳基或烷基之外的取代基时, 会出现大量的脱甲氧基副产物18.其反应机理如Scheme 5所示.此外, 经酯与内酯(path d)、酰亚胺(path e)、混酐(path f)及醛(path g)等为原料与DMHA缩合, 均可制备WAs[6], 近几年报道不多, 代表性例子[24~27]可见Scheme 6.近些年, 过渡金属Pd催化合成乙烯基或芳基WAs也有报道(path h 和i), 主要包括以下两种方法: (1)在Heck反应条件下, DMHA 和CO 可分别与芳溴31[28]、内酰胺/内酯的三氟甲磺酸酯32[29]进行甲酰胺化反应, 制备WAs (Eq. 4). 2011年, Wieckowska等[30]对上述反应进行了改进, 采用W(CO)6作为固体CO源, 代替之前往体系中不断通入CO气体, 成功地对芳溴和芳碘进行了甲酰胺化反应, 但反应中存在N—O键断裂的副反应. 此外, 由于芳碘比芳溴更活泼, 可用芳碘代替芳溴作为反应底物, 采用PPh3与Pd(OAc)2配合, 避开使用价格昂贵的含磷配体Xantphos. 可能的反应机理如Scheme 7, PPh3 将Pd还原为0价络合物35, 芳碘与35氧化加成形成中间体36, 后者再激活CO得到末端羰基络合物37, 随后进行CO的插入反应及碱性条件下的还原消除, 得到终产物WAs及Pd络合物35[31]. (2)乙烯基或芳基取代的锡烷39[32]或硼酸40[33]作反应底物, 与甲酰氯41进行Stille-type 交叉偶联反应制备WAs (Eq. 5), 该方法可以顺利制备α,β-不饱和WAs.对于α,β-不饱和WAs的制备, 既可采用传统地缩合成酰胺方法, 通过α,β-不饱和羧酸与DMHA制备, 也可通过Pd催化Stille-type交叉偶联反应制备, 这两种途径在前面已经提到. 另外, 还可以通过各种醛进行Wittig反应[34]、HornerWadsworth-Emmons反应[35]及Julia 成烯反应[36]等制备, 其反应中间体分别为: 44, 45~49, 50~52 (Scheme 8).44参与的Wittig反应均生成E式α,β-不饱和WAs产物, 而Horner-Wadsworth-Emmons反应中, 46, 47及49能控制反应主要生成Z式α,β-不饱和WAs产物. 2012年, Yamada等[35f]利用45经Horner-Wadsworth-Emmons反应得到53, 后者作为新的HWE反应合成砌块, 与醛经多次HWE反应成功合成了花药黄素54(Scheme 9).在Julia成烯反应中, 50[36a]及52[36b]均与各种醛得到E式α,β-不饱和WAs产物. 含氟化合物51在不同反应体系下可以进行立体化学控制. 比如: 在含碳酸钾的DMF中, 51与醛室温反应主要得到Z式产物[36c]; 而在THF溶剂体系中, NaH 作为碱可得Z式产物收率大于98%; 而DBU作为碱可通过控制溶剂极性得到Z式或E式α,β-不饱和WAs作为主要产物[36d]. 另外, 室温搅拌下, 在CsCO3作为碱的二氯甲烷溶液中, 过量多聚甲醛可与51顺利得到含氟端烯WAs, 后者并不涉及顺反异构[36e]. 2014年, Chowdhury等[36f]报道了以51为原料, 经两次Julia成烯反应合成氟代共轭二烯WAs 57 (Scheme 10).最近, Aidhen等[37,38]利用含苯环WAs合成子58与醛进行Julia 成烯合成了末端含WAs的二苯乙烯化合物. 2014年, 他们组[39]还利用59与多聚甲醛Julia成烯反应, 在苯甲酰胺的α位引入乙烯基, 氧化双键后可制备α-乙酰基二苯甲酮类化合物.Weinreb酰胺在合成上的应用是非常广泛的, 包括构建各种合成等价体与合成砌块、杂环、天然产物, 甚至在医药中间体大规模生产[6]等领域, 均有其大量应用. 下面总结了近五年来, Weinreb 酰胺参与的有机合成反应.2.1 成醛酮反应Weinreb酰胺在合成中最重要的应用就是通过它与各种金属有机试剂反应得到结构各异的醛酮, 后者再经一系列变化, 可用于各种复杂结构分子的合成. WAs 的最大特点是, 反应过程中金属离子可与WAs 的羰基氧和甲氧基氧形成稳定的螯合环, 因此不会与过量金属试剂发生过度加成, 从而保证了反应的清洁性. 现今, 此方法已经成为由羧基转化为醛酮最可靠的方法.一般地, 铝试剂可以将WAs还原为醛[40]; 格氏试剂或锂试剂与WAs反应, 可以制备脂肪酮[41]、烯酮[42]、芳香酮[43]及炔酮[44]等, 锂试剂反应活性比格氏试剂高, 一般在更低温下进行反应, 这是WAs最常见的成酮方法(Scheme 11).2010年, Aidhen等[47]报道了含叶立德Weinreb酰胺67先与各种单糖进行wittig反应构建C—C键68, 再将WAs基团与各种芳基格氏试剂反应, 得到单糖修饰的二苯酮衍生物, 即Phenstatin衍生物69. 后者具有重要的抗癌活性(Scheme 14). 之前, 有人报道过利用Wittig试剂与WAs的羰基进行反应, 可以合成酮[48]. 但在此反应条件下, 67的WAs基团并不参与wittig反应.Weinreb酰胺不但可以用于制备普通酮, 还可以制备α 卤代酮. 2012 年,Leadbeater 等[63]报道了利用Rupper-Prakash试剂89与WAs88反应合成三氟甲基酮90 (Eq. 6). 但底物为α,β-不饱和WAs 时, 在该反应条件下会出现迈克尔加成副产物, 即N-甲基-N-甲氧基氨基负离子会进攻双键, 会严重影响三氟甲基酮的产率.2.2 1,4-共轭加成反应α,β-不饱和WAs作为重要的有机合成中间体, 其反应活性主要体现在羰基和双键两个官能团上. 有机金属试剂可与其羰基进行成酮反应, 这在上一节中已经提到. 这里我们介绍其1,4-共轭加成反应[69](Scheme 21).2008年, Olivella等[69a]报道了TiCl4催化乙醇酸衍生物99与N-甲基-N-甲氧基丙烯酰胺100的1,4-共轭加成反应(Scheme 22).2.3 烯烃复分解反应在氯仿中, 己二烯与含WAs基团烯烃在Grubbs二代催化剂钌卡宾配合物111催化下, 可顺利进行双向复分解反应[69c], 微波加热可加速反应进行. 该类金属卡宾催化剂可利用异丙氧基苯乙烯作为配体, 进一步替代卡宾配合物111中的膦配体而提高催化活性. Lee 等[72]发现利用Grubbs二代改良型催化剂112, 烯丙基卤113与α-烷氧基WAs 114可顺利进行复分解反应. 烯基与WAs基的距离并不影响反应收率, 但当用NH(CH3)2代替WAs, 反应几乎不能发生. 当使用116作为反应底物时, 双键与WAs基团距离过近, 会形成稳定的中间体117或118, 不利于烯烃复分解反应的进行.2.4 烯醇式亲核反应α-氨基酸及其衍生物在化学和生物学中扮演着重要角色, 它是构成肽和蛋白质的单体. α-氨基酸在有机化学中也有广泛应用, 比如在不对称合成中可作为重要模板、在全合成中充当合成砌块以及在天然活性物质中常作为亚结构出现等. 近几年, 利用烯醇式WAs酰胺119与手性亚胺120进行亲核加成, 合成手性β-氨基酸衍生物121被报道(Scheme 26). 比如N-膦酰基122[73]和N-手性亚砜123[74,75]均可作为手性辅助基团, 不对称催化合成手性β-氨基酸WAs, 随后可用强酸脱掉这些辅助基团.2.5 C—H活化反应2012年, Zhang等[79]报道了利用Rh或Ru催化丙烯酰胺与烯烃的脱氢偶联反应, 用于合成(Z,E)-二烯酰胺化合物. 其中, 133 作为烯烃底物也能很好的适用于该反应, 反应收率中等, Z/E>98/2 (Eq. 8).2.6 催化氢化反应2013年, Kumaraswamy 等[86]报道了利用过渡金属Ru催化不对称转移氢化反应, 对α-烷基取代-β-酮Weinreb酰胺145进行动态动力学拆分, 即DKR-ATH(dynamic kinetic resolution-asymmetric transfer hydrogenation)反应. 该反应可一步得到两个连续的手性中心、立体构型明确的产物分子146 (Eq. 12). 146可作为重要中间体, 经多步反应全合成天然产物(-)-brevis- amide及其对映异构体.2.7 关环反应Weinreb酰胺也常参与杂环的合成. 通过调研文献发现, WAs可作为分子内亲电基团应用于Parham 环化反应, 反应中生成的芳基锂153[90]进攻分子内的WAs, 形成关环产物.2.8 其他应用Evans 等[93]在2010 年曾经将WAs基团引入Jones-Moss非氮卡宾前体161, 通过光照得到卡宾162.Weinreb酰胺虽然在有机合成上取得巨大成功, 但值得注意的是, 在一些反应中已经出现Weinreb酰胺中的N—O键发生断裂的现象, 即脱甲氧基副反应, 导致WAs分解. 最早报道此现象的是Graham 等[94], 他们发现在-78 ℃下, WA 163与强碱LDA反应会发生脱甲氧基, 形成164为主要产物. 这个分解反应可能是一个释放甲醛的E2消除机理(Eq. 16).Weinreb酰胺脱甲氧基现象的报道, 在Labeeuw早期发表的文章[95]前言中已有总结. 在该文中, 他们也发现WAs 165, 无论如何改变反应温度、溶剂及有机锂试剂的摩尔量, 主产物不是酮167, 而是脱甲氧基产物168. 当用叔丁基166代替甲基后, 脱甲氧基副反应能被很好地抑制, 成酮产物167收率迅速提高到72%, 但由于位阻增大, 反应时间会增长(Eq. 17). 另外, Li/ DTBB[96]和LiSnBu3[97]也可使WAs发生脱甲氧基反应.上述WAs发生脱甲氧基副反应, 均在有机锂及格氏试剂等强碱环境下发生. 在WAs参与的过渡金属催化反应中也会偶尔发生脱甲氧基副反应, 比如前面提到的Pd催化偶联[30]及Ru催化氢化[87]等反应. 2011 年, Fukuzawa等[98]专门研究了Ru催化下的WAs N—O键断裂反应. 该反应不需要任何有机配体, RuCl3与还原剂Cu-Zn配合使用, 可在甲醇中将烷基型、乙烯型及芳香型WAs顺利脱甲氧(Eq. 18). 反应机理涉及Ru的氧化还原催化循环.另外, 非金属的电中性有机超电子给体171[99]和172[100]利用单电子转移过程, 通过自由基机理也可使Weinreb酰胺的N—O键发生断裂.综上所述, Weinreb酰胺因其易制备、便于储藏及特殊的反应性质, 在有机合成中已得到广泛关注. 它既可作为酰化试剂与有机金属试剂反应, 而不会过度加成; 又可作为羰基的等价体参与许多类型反应, 起到保护羰基的作用, 从而表现出很好的官能团耐受性. 鉴于其可靠的成醛酮反应性质, Weinreb 酰胺已在天然产物等各种复杂体系的合成中发挥重要作用, 并成功应用于工业大规模生产中. 然而, Weinreb 酰胺在使用中也暴露出一些问题, 比如: 由于普遍使用有机金属试剂与Weinreb酰胺进行成酮反应, 此苛刻反应条件会限制酰胺底物中其它官能团的多样性; 个别反应条件下, 其N—O键断裂作为副反应会导致自身分解; N-甲氧基-N-甲基基团结构过于简单, 缺乏对反应的立体控制等. 今后, 为了克服上述问题, 寻求温和反应条件提高选择性、探索降低副产物的合成方法及开发Weinreb酰胺手性替代基团将成为又一研究热点[7]. 与此同时, 坚持开发简单高效的Weinreb酰胺合成方法, 探索Weinreb酰胺作为合成砌块或等价体在不同反应体系中的官能团耐受性还将持续下去, 为其在更广泛领域的应用提供保障.声明:。

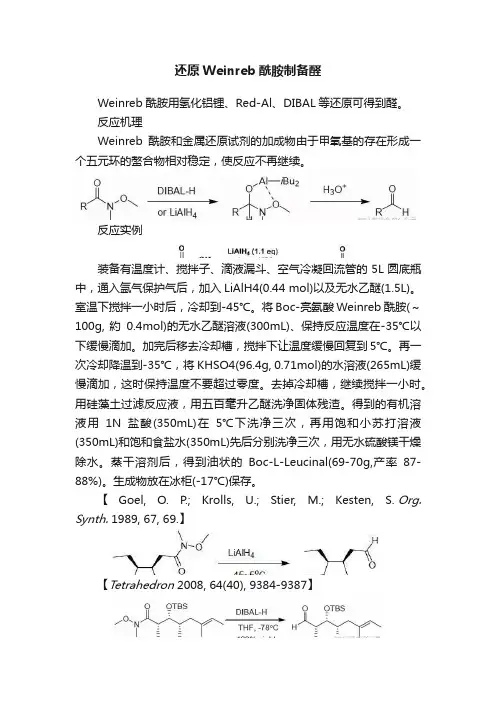
还原Weinreb酰胺制备醛Weinreb酰胺用氢化铝锂、Red-Al、DIBAL等还原可得到醛。
反应机理Weinreb酰胺和金属还原试剂的加成物由于甲氧基的存在形成一个五元环的螯合物相对稳定,使反应不再继续。
反应实例装备有温度计、搅拌子、滴液漏斗、空气冷凝回流管的5L圆底瓶中,通入氩气保护气后,加入LiAlH4(0.44 mol)以及无水乙醚(1.5L)。
室温下搅拌一小时后,冷却到-45℃。
将Boc-亮氨酸Weinreb酰胺(~100g, 約0.4mol)的无水乙醚溶液(300mL)、保持反应温度在-35℃以下缓慢滴加。
加完后移去冷却槽,搅拌下让温度缓慢回复到5℃。
再一次冷却降温到-35℃,将KHSO4(96.4g, 0.71mol)的水溶液(265mL)缓慢滴加,这时保持温度不要超过零度。
去掉冷却槽,继续搅拌一小时。
用硅藻土过滤反应液,用五百毫升乙醚洗净固体残渣。
得到的有机溶液用1N盐酸(350mL)在5℃下洗净三次,再用饱和小苏打溶液(350mL)和饱和食盐水(350mL)先后分别洗净三次,用无水硫酸镁干燥除水。
蒸干溶剂后,得到油状的Boc-L-Leucinal(69-70g,产率87-88%)。
生成物放在冰柜(-17℃)保存。
【 Goel, O. P.; Krolls, U.; Stier, M.; Kesten, S. Org. Synth. 1989, 67, 69.】【Tetrahedron 2008, 64(40), 9384-9387】【Angew. Chem. Int. Ed. 2007, 119(45), 8862-8865】4-[4-(methanesulfonamido)phenyl]butyraldehydeA mixture of 4.20 g (14 mmol) of4-[4-(methanesulfonamido)phenyl]butyric acid, N-methoxy-N-methylamide and 100 mLof anhydrous tetrahydrofuran was stirred under nitrogen with cooling in an icebath as 17.5 mL (17.5 mmol) of 1Mlithium aluminum hydride in tetrahydrofuran was added gradually by syringe. After 0.75 hours, 70 mL of 5percent potassiumhydrogen sulfate solution (aqueous) was added cautiously by syringe. Themixture was then removed from the ice bath, diluted with 150 ML of water, andshaken with 150 mL of ethyl acetate. The milky aqueous phase was extracted withan additional 50 mL of ethyl acetate. The combined organic fractions werewashed successively with 2*100 mL of 1N hydrochloric acid, then 50 ML ofsaturated aqueous sodium bicarbonate solution, and finally 50 ML of saturatedaqueous sodium chloride solution. The organic phase was dried over magnesiumsulfate, filtered, and concentrated in vacuo. Flash chromatography of theresidue on silica gel (elution with 3:2 hexane-EtOAc) yielded 2.47 g (73%) of an oil; homogeneous by TLC in1:1 hexane-EtOAc).Upon storage in the freezer, solidification occurred (mp 41~44oC.).【US5756507】。
经典化学合成反应标准操作酰胺及酰亚胺的合成目录1. 前言 (2)2. 羧酸与胺的缩合酰化反应 (2)2.1活性酯法 (2)2.1.1应用氯甲酸乙酯或异丁酯活性酯法合成酰胺示例 (4)2.1.2应用氯甲酸乙酯或异丁酯活性酯法合成伯酰胺示例 (4)2.1.3应用羰基二咪唑合成Weinreb酰胺示例 (5)2.1.4应用的磺酰氯合成酰胺示例 (5)2.1.5应用Boc酸酐合成伯酰胺示例 (6)2.2碳二亚胺类缩合剂法 (6)2.2.1应用DCC缩合法合成酰胺示例 (8)2.2.2应用DIC缩合法合成酰胺示例 (9)2.2.3应用EDC缩合法合成酰胺示例一(二氯甲烷为溶剂) (9)2.2.4应用EDC缩合法合成酰胺示例二(DMF为溶剂) (10)2.3 鎓盐类的缩合剂法 (10)2.3.1应用HATU/TBTU为缩合剂合成酰胺示例 (12)2.3.2应用BOP为缩合剂合成酰胺示例 (13)2.3.3应用PyBOP为缩合剂合成酰胺示例一(常规) (13)2.3.4应用PyBOP为缩合剂合成酰胺示例二(用于合成伯酰胺) (14)2.4 有机磷类缩合剂 (14)2.4.1应用DPP-Cl为缩合剂合成酰胺示例 (15)2.4.2应用DPPA为缩合剂合成酰胺示例 (15)2.4.3应用BOP-Cl为缩合剂合成酰胺示例 (16)2.5.1应用三苯基磷-多卤代甲烷合成酰胺示例 (17)2.5.2应用三苯基磷-六氯丙酮合成酰胺示例 (17)2.5.3应用三苯基磷-NBS合成酰胺示例 (18)3. 氨或胺与酰卤的酰化反应 (18)3.1酰卤的制备示例 (19)3.5.1应用二氯亚砜合成酰氯示例 (19)3.5.2用草酰氯合成酰氯示例 (20)3.5.3用三氯均三嗪合成酰氯示例 (20)3.5.4用三氟均三嗪合成酰氟示例 (21)3.1应用酰卤的合成酰胺 (21)3.5.1应用酰氯合成酰胺示例(有机碱) (21)3.5.2应用酰氯合成酰胺示例(无机碱) (21)3.5.3应用酰氟合成酰胺示例 (23)4. 氨或胺与酸酐的酰化反应 (23)4.2酸酐合成酰胺示例 (24)5. 其他缩合方法 (24)1. 前言酰胺化是有机合成中最基本,也是最重要的合成方法之一。
weinerb 酰胺反应机理概述及解释说明1. 引言1.1 概述Weinerb酰胺反应是一种重要的有机合成反应,广泛应用于药物合成、材料科学和生物化学等领域。
该反应以亚磷酸盐为催化剂,在适当的条件下,通过吡咯烷酮与羧酸衍生物的反应生成相应的酰胺产物。
这一反应机理的探索对于揭示有机反应的原理以及开发新型催化剂具有重要意义。
1.2 文章结构本文将从以下几个方面对Weinerb酰胺反应进行概述和解释说明:- 引言:介绍文章内容、目的和结构;- Weinerb酰胺反应机理概述:简要介绍该反应的基本情况,包括反应简介、基本步骤和反应条件;- Weinerb酰胺反应机理解释说明:详细讨论该反应的关键步骤,包括中间体形成、催化剂作用机制以及副产物生成及影响因素;- 应用领域与未来展望:总结该反应在工业上的实际应用,并展望其未来的研究进展和挑战,并提出发展方向建议;- 结论:总结回顾本文的内容,强调该反应的研究价值和意义,并给出对未来研究的建议。
1.3 目的本文旨在全面了解Weinerb酰胺反应的机理,深入探讨其中关键步骤的解释说明。
通过文章阐述,希望能够提供有关这一反应的详尽知识,并为其在实际应用和未来研究方向上提供参考。
2. Weinerb 酰胺反应机理概述:2.1 反应简介Weinerb酰胺反应是一种重要的有机合成方法,通常用于合成酰胺化合物。
该反应以底物中的羧酸和胺为原料,在适当的条件下经过一系列步骤进行,最终生成相应的酰胺产物。
这个反应具有高效、选择性好以及广泛的底物适用性等优点,因此在有机合成领域被广泛应用。
2.2 基本步骤Weinerb酰胺反应涉及三个基本步骤:缩水、缩聚和水解。
首先是缩水步骤,也称为活化步骤。
在这一步中,羧酸底物与缩水剂(例如二甲基亚砜或1-甲基咪唑)反应形成临时结构活化羧酸中间体。
这个中间体对后续反应步骤非常关键。
接下来是缩聚步骤,即活化羧酸与胺底物之间的偶联反应。
通过加入碱性氢氧化物作为催化剂,并控制适当的温度和时间,活化的羧酸中间体与胺底物发生缩聚反应,生成酰胺产物。
关于Weinreb酰胺
N-甲基-N-甲氧基酰胺,又称Weinreb酰胺(WAs),是一种被广泛应用于有机合成中的酰基化试剂。
Weinreb酰胺既可与格氏试剂或有机锂试剂反应合成酮,也可经金属氢化物还原成醛,且金属试剂过量不会导致产品过度加成。
此外,Weinreb酰胺具有易制备、稳定易存储,可放大反应的优势。
制备
Weinreb酰胺一般可通过羧酸活化、羧酸衍生物(酰卤、酯、内酯、酰亚胺、酸酐、醛等)与N,O-二甲基羟基胺盐酸盐(DMHA)缩合、三氯甲基醇经同系化-胺化反应等制备。
近年来,也有报道,过渡金属Pd催化合成反应也可合成乙烯基或芳基WAs。
主要反应
Weinreb酰胺在合成中最重要的应用就是通过WAs能与各种金属有机试剂反应得到醛酮,且该反应中金属离子可与WAs的羰基氧和甲氧基氧形成稳定的螯合环,因此不会发生过度加成(反应机理详见下图)。
其中,Weinreb 酰胺还能参与Wittig反应和Shapiro反应制得醛酮。
此外,在一些反应中,WAs会发生脱甲氧基副反应,即N-O键发生断裂的现象,导致WAs分解。
常用Weinreb酰胺。
常用试剂----Weinreb酰胺
图片链接:/2018/06/17/4-weinreb-amide-1981/
N-甲氧基-N-甲基酰胺俗称Weinreb酰胺、它能与Grignard试剂或有机锂试剂反应生成酮。
酰卤或是酯中加入两倍当量的格式试剂或是有机锂试剂的话会得到醇,而Weinreb酰胺则能够避免这种过度的加成。
Weinreb酰胺和金属还原试剂的加成物由于甲氧基的存在形成一个五元环的螯合物相对稳定,使反应不再继续。
Grignard试剂或有机锂试剂反应生成酮,也类似。
Weinreb酰胺的合成方法见上图,主要分为以下几种:1、由酸直接缩合制备得到;2、由酯进行胺酯交换制备得到,3、由酰氯或酸酐直接反应得到。
具体方法参考:酰胺的制备。
强碱性条件下,E2消除,可能会导致副反应,影响产率,叔丁基醚可以避免此副反应。
图片来源:【EOC化学资讯】 Weinreb酰胺在有机合成中的应用试剂应用(详细内容点击查看)
一、还原Weinreb酰胺制备醛
二、weinreb酰胺制备酮
三、Shapiro reaction
四、Wittig试剂和weinreb酰胺反应
五、作为导向基参与C-H键活化反应【C-H活化反应】
【EOC化学资讯】 Weinreb酰胺在有机合成中的应用。
矿产资源开发利用方案编写内容要求及审查大纲
矿产资源开发利用方案编写内容要求及《矿产资源开发利用方案》审查大纲一、概述
㈠矿区位置、隶属关系和企业性质。
如为改扩建矿山, 应说明矿山现状、
特点及存在的主要问题。
㈡编制依据
(1简述项目前期工作进展情况及与有关方面对项目的意向性协议情况。
(2 列出开发利用方案编制所依据的主要基础性资料的名称。
如经储量管理部门认定的矿区地质勘探报告、选矿试验报告、加工利用试验报告、工程地质初评资料、矿区水文资料和供水资料等。
对改、扩建矿山应有生产实际资料, 如矿山总平面现状图、矿床开拓系统图、采场现状图和主要采选设备清单等。
二、矿产品需求现状和预测
㈠该矿产在国内需求情况和市场供应情况
1、矿产品现状及加工利用趋向。
2、国内近、远期的需求量及主要销向预测。
㈡产品价格分析
1、国内矿产品价格现状。
2、矿产品价格稳定性及变化趋势。
三、矿产资源概况
㈠矿区总体概况
1、矿区总体规划情况。
2、矿区矿产资源概况。
3、该设计与矿区总体开发的关系。
㈡该设计项目的资源概况
1、矿床地质及构造特征。
2、矿床开采技术条件及水文地质条件。