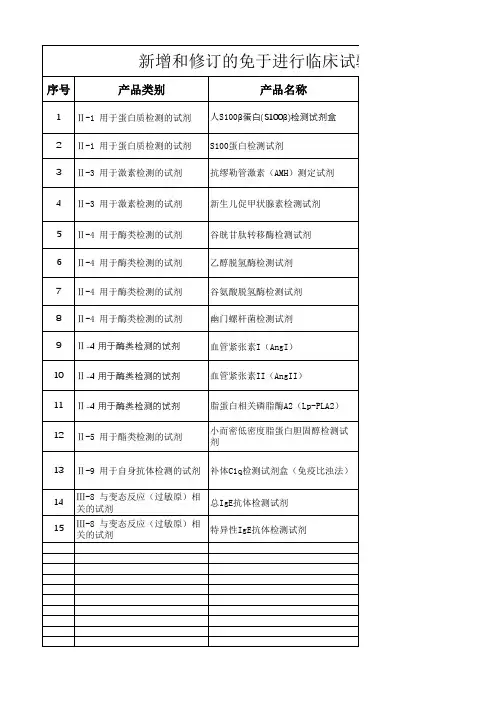
《隐球菌抗原检测试剂注册技术审查指导原则(征求意见稿)》
- 格式:doc
- 大小:111.90 KB
- 文档页数:25
— 1 — 隐球菌抗原检测试剂 注册技术审查指导原则
(征求意见稿)
本指导原则旨在指导注册申请人对隐球菌抗原检测试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。 本指导原则是对隐球菌抗原检测试剂的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。 本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但需要提供详细的研究资料和验证资料,相关人员应在遵循相关法规的前提下使用本指导原则。 本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时进行调整。 一、范围 隐球菌病是一种由隐球菌感染引起的深部真菌病,可累及全身多个系统。隐球菌属(Cryptococcus,有性期称线黑粉菌属)— 2 —
隶属于真菌界,双核亚界、担子菌门、伞菌亚门、银耳纲、银耳目、银耳科,有性期隶属于担子菌门、线黑粉菌目、线黑粉菌科。属内包括近70个种,其中对人致病的最主要病原菌是新生隐球菌(Cryptococcus neoformans)和格特隐球菌(Cryptococcus gattii)。根据新生隐球菌荚膜多糖成分和生化方面的差异,将
新生隐球菌分成2个变种(新生隐球菌新生变种C.neoformans var.neoformans和新生隐球菌格鲁比变种C.neoformans var.grubii),按血清学分为A、B、C、D和AD型5个型。其中新
生变种为血清D、AD型,欧洲、南美洲流行;格鲁比变种为血清A型,全球流行;新生隐球菌为血清B、C型,在热带\亚热带\温带流行;我国血清型主要为A型。此外,还发现了新生变种与格鲁比变种的杂合体(血清型AD)。隐球菌的基因型包括新生隐球菌的VNⅠ、Ⅱ、Ⅳ、B 和格特隐球菌的VGI-VGIV。两种隐球菌的无性繁殖体均为无菌丝的单芽孢酵母菌,在体外为无荚膜或仅有小荚膜, 进入人体内后很快形成厚荚膜, 有荚膜的隐球菌菌体直径明显增加,致病力明显增强。隐球菌荚膜的主要成分荚膜多糖是确定血清型特异性的抗原基础,并与其毒力、致病性及免疫性密切相关。隐球菌是一种机会感染性真菌,主要侵犯免疫力低下及免疫缺陷人群,也可发生于免疫正常者,可以感染人体的任何组织和脏器,最常见的部位是中枢神经系统,其次是肺部和皮肤。目前, 在免疫抑制患者中, 隐球菌感染的发病率约为5%~10%,— 3 —
在AIDS患者中, 隐球菌的感染率可以高达30%;而在免疫功能正常的人群中, 隐球菌的感染率约为十万分之一左右。 中枢神经系统隐球菌感染通常表现为脑膜炎或脑膜脑炎, 极少数表现为单个或多个局灶性肿块损害(隐球菌肉芽肿),中枢神经系统感染可伴发肺部或其他播散性感染, 但大多数不伴有其他感染的临床表现。通过对脑脊液样本进行培养、墨汁染色涂片可明确中枢神经系统感染诊断。 隐球菌感染中枢神经系统外的器官和部位,肺和皮肤是常见的两个部位。肺是隐球菌感染的主要门户,肺隐球菌感染的临床表现多种多样, 可从无症状的结节到严重的急性呼吸窘迫综合征(ARDS)。皮肤隐球菌感染根据感染来源, 可以分为原发性和继发性感染两种。继发性隐球菌感染一般预示已经发生播散性隐球菌感染, 主要来源于血行播散, 提示感染严重。确诊主要依靠组织病理检查和病原学涂片和培养。血清隐球菌荚膜多糖抗原检测阳性有临床疑似诊断价值。 隐球菌感染的病原学及实验室检测包括隐球菌的微生物学鉴定,隐球菌病的免疫学诊断(血清学实验),隐球菌的药物敏感试验和隐球菌病的组织病理学。其中隐球菌病的免疫学诊断(血清学实验)临床上常用的为隐球菌荚膜抗原检测,其方法有乳胶凝集试验(latex agglutination test ,LA) 、酶联免疫分析法(enzyme immunoassay ,EIA)及侧流免疫层析法(lateral — 4 —
flow immunoassay ,LFA)等。隐球菌荚膜抗原检测可辅助诊断隐球菌感染,隐球菌荚膜多糖抗原阳性提示隐球菌感染,脑脊液滴度的高低可提示疾病的严重程度。隐球菌荚膜抗原检测因其简单、快速已经是目前国内临床辅助诊断隐球菌感染最常用的方法之一。 本指导原则所指隐球菌抗原为隐球菌荚膜多糖抗原,适用于利用酶联免疫法、乳胶凝集试验和侧向免疫层析法等基于抗原抗体反应原理,对来源于血清和/或血浆和/或全血和/或脑脊液等人体样本中的隐球菌荚膜多糖抗原进行体外定性检测的试剂。结合临床和其他实验室指标,可用于隐球菌感染的辅助诊断。如涉及半定量检测试剂,或待测样本类型为血清、血浆、全血、脑脊液外其他样本类型的(如尿液、肺泡灌洗液),应根据申请的预期用途设计临床试验进行验证。可适量参考本指导原则相关要求。 隐球菌抗原检测的准确性至关重要,不正确的检测结果可能导致对患者管理决策失误,假阴性结果可能导致诊断不及时而延误治疗,假阳性结果可能导致不必要的干预措施。申请人应建立良好的产品性能,并对安全性和有效性进行科学合理的验证。 指导原则仅包括对隐球菌抗原检测试剂申报资料中部分项目的要求,适用于进行产品注册和相关许可事项变更的产品。其他未尽事宜,应当符合《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)(以下简称《办法》)、《体外诊断试剂注册申报资料基本要求》(国家食品药品监督管理总局公告2014年第44号)等相关法规和文件的要求。 — 5 —
二、注册申报资料要求 (一)综述资料 综述资料主要包括产品预期用途、产品描述、有关生物安全性的说明、研究结果的总结评价以及同类产品上市情况介绍等内容。其中,需注意以下内容: 1.产品预期用途 详述检测试剂的预期用途,包括预期人群、适用的样本类型,检测结果的临床指导意义等。描述隐球菌的生物学特征、隐球菌感染疾病相关流行病学、潜伏期、易感人群、临床表现等。说明现有的临床或其他实验室诊断方法等。 2.同类产品上市情况 应着重从技术方法及预期用途(包括预期人群、样本类型、临床意义)等方面写明拟申报产品与现行临床诊断方法,以及目前市场上已获批准的同类产品之间的主要异同点。 (二)主要原材料研究资料 此产品的主要原材料包括抗体、固相载体、质控品(如适用)以及企业参考品等。应提供主要原材料的选择与来源、制备过程、质量标准等研究资料。如主要原材料为企业自制,应明确原材料的制备原理,简述制备过程,其生产工艺必须相对稳定;如主要原材料为外购,应提供的资料包括:原材料的筛选研究资料、供应商选择的研究资料、质量标准、供应商出具的主要原材料检验报告(质量证书)和入厂检验报告等。供应商为原材料的生产商,不得随意更换。 主要原材料的筛选研究资料,应包含试验方法、实验数据及— 6 —
分析。质量控制标准应包括相应试验方法及原材料应达到的客观评价标准。人源或其他生物源性原材料质量控制标准中还应包含生物安全指标。生产企业应依据质量控制标准对原材料进行检验,检验合格方可用于生产。 1. 抗体 应明确抗体名称、生物学来源、免疫原、供应商名称、质量标准(一般包括外观、蛋白纯度、浓度、效价、分子量、功能性试验)等。 如为自制抗体,还需明确抗体所针对的抗原表位、免疫原的具体信息,包括天然抗原的来源、重组或人工合成抗原的序列信息及制备方法。同时提交抗体制备、筛选、鉴定等详细试验资料。 2. 其他主要原辅料 包括基于酶联免疫技术的固相载体,如酶标板、微粒等;基于凝集反应的乳胶颗粒等;基于侧向免疫层析技术的金颗粒、硝酸纤维素膜等。申请人应提交上述原材料的选择依据及验证资料,明确供应商名称及原材料质量标准。 3.试剂盒质控品(如有) 应包括原料来源、质控品制备、阴阳性确认和质量标准等相关研究资料。质控品应至少包含阴性和阳性两个水平。阳性质控品可选择标准菌株或经鉴定、可溯源的临床分离株或荚膜多糖抗原。如荚膜多糖抗原为企业自制,请详述制备的方法。提交质量标准中各项指标,如浓度、纯度、活性测定的方法及结果。 4.企业参考品 如申报产品有相应的国家参考品,则企业参考品应参考国家— 7 —
参考品的项目设置,应不低于国家参考品要求。若尚无国家参考品,申请人应根据产品性能验证的实际情况自行设定企业参考品。 应提交企业参考品的原料选择、制备、阴阳性及浓度/滴度确认等相关验证资料。说明参考品阴阳性及浓度/滴度确认的方法或试剂(建议采用国内已上市的、临床上普遍认为质量较好的同类试剂)。参考品可采用临床真实样本,亦可采用标准菌株或临床分离株添加入待测样本基质的形式制备。企业参考品的基质应与待测样本相同。企业参考品的设置建议如下: 4.1阳性参考品 阳性参考品应考虑覆盖不同种及不同型别的隐球菌感染样本。建议包含新生隐球菌VNⅠ、Ⅱ、Ⅳ、B基因型和格特隐球菌VGI-VGIV基因型。参考品应设置不同浓度水平。 4.2 阴性参考品 阴性参考品应考虑检测特异性的评价,纳入正常临床样本、含类风湿因子等干扰因素的样本及其他病原体阳性样本,建议包含其他常见酵母菌和霉菌、HIV等。 4.3检测限参考品 可设置不同滴度的临床阳性样本/荚膜多糖抗原或临床阳性样本的系列梯度浓度样本,样本基质与检测样本一致,其中应包含检测限水平。 4.4精密度参考品 建议至少包含中阳性和弱阳性,至少两个浓度水平的参考品。