FDA批准的激酶小分子抑制剂类药物及分类一览
- 格式:doc
- 大小:3.09 MB
- 文档页数:13

网络出版时间:2023-08-2809:25:34 网络出版地址:https://link.cnki.net/urlid/34.1086.r.20230825.1002.006酪氨酸激酶抑制剂引起的肝损伤机制研究进展刘慧慧,魏静瑶,张丽珍,冯进伟,刘瑞娟,田 鑫(郑州大学第一附属医院药学部,河南郑州 450052)收稿日期:2022-03-17,修回日期:2022-06-21基金项目:国家自然科学基金资助项目(No81903720)作者简介:刘慧慧(1998-),女,硕士生,研究方向:药理学,E mail:lhh18538277781@163.com;刘瑞娟(1988-),女,博士,副主任药师,研究方向:临床药理学,通信作者,E mail:fccliurj@zzu.edu.cn;田 鑫(1975-),女,博士,教授,博士生导师,研究方向:药理学,通信作者,E mail:tianx@zzu.edu.cndoi:10.12360/CPB202203052文献标志码:A文章编号:1001-1978(2023)09-1613-05中国图书分类号:R 05;R345 57;R575;R977 3摘要:酪氨酸激酶抑制剂(tyrosinekinaseinhibitors,TKIs)为一类靶向抑癌基因相关受体酪氨酸激酶的小分子化合物,通过阻断下游的信号通路发挥抗癌作用。
TKIs广泛用于癌症的治疗,对于部分肿瘤显示出较传统化疗药物更好的疗效。
然而,TKIs引起的药物性肝损伤是其在临床应用中面临的难题之一。
笔者通过查阅国内外相关文献,对TKIs的分类、临床应用及其引起肝损伤的机制等进行综述,以期为阐明TKIs肝损伤的机制和寻找有效的防治手段提供一定的参考。
关键词:酪氨酸激酶;酪氨酸激酶抑制剂;药物性肝损伤;靶向药物;机制;靶点开放科学(资源服务)标识码(OSID): 酪氨酸激酶(tyrosinekinases,TKs)对于肿瘤细胞的信号转导、细胞增殖、转移和凋亡发挥着重要作用[1],以TKs作为靶点进行相关药物研发是当前抗肿瘤药物研究的热点。

ALK(酪氨酸激酶)是一种蛋白质,可以参与肿瘤的生长和转移。
针对ALK突变的肿瘤,已经开发出了多种靶向药物。
根据药物的开发历史和临床应用情况,ALK靶点药物一般分为一、二、三代三个代别。
一代ALK靶点药物:一代ALK抑制剂是最早出现的药物,如克唑替尼(Crizotinib)。
这类药物的作用机制是抑制ALK的酪氨酸激酶活性,从而阻断肿瘤细胞的生长和扩散。
但是,这些药物存在一定的耐药性,并且不能有效地穿过血脑屏障,因此对于存在脑转移的肿瘤治疗效果不佳。
二代ALK靶点药物:二代ALK抑制剂是在一代药物基础上进行改良的药物。
例如,里拉替尼(Lorlatinib)、布里加丙胺(Brigatinib)等。
这些药物可以抑制ALK的酪氨酸激酶活性,并且具有更强的候选性和更好的耐药性。
此外,这些药物可以穿过血脑屏障,并对存在脑转移的肿瘤具有更好的治疗效果。
三代ALK靶点药物:三代ALK抑制剂是最新开发的一类药物,例如沙可维替尼(Selpercatinib)。
这些药物不仅可以抑制ALK的酪氨酸激酶活性,还可以针对其他突变,如RET、ROS1等进行干预。
这些药物具有更高的选择性和更好的安全性,同时能够有效地穿过血脑屏障,对于存在脑转移的肿瘤具有较好的治疗效果。
总的来说,ALK靶点药物的一、二、三代分类主要根据药物的开发历史、作用机制、耐药性和临床应用效果等因素进行划分。
每一代药物都有其特点和优缺点,医生可以根据患者的具体情况进行选择和应用。


alk抑制剂一代二代产品的划分原则ALK(Anaplastic lymphoma kinase)抑制剂是一类针对ALK基因突变引起的肿瘤进行治疗的药物。
根据研发时间和药物特性的不同,ALK抑制剂可以被划分为一代和二代产品。
下面将介绍ALK抑制剂一代和二代产品的划分原则。
一代ALK抑制剂是指最早出现的ALK抑制剂药物,也是最早用于临床治疗的药物。
一代ALK抑制剂包括Crizotinib(金翎唑)、Ceritinib(富罗达胺)、Alectinib(艾达宁)等。
这些药物主要通过抑制ALK蛋白的酪氨酸激酶活性来阻断肿瘤生长和扩散。
一代ALK抑制剂的初始疗效非常显著,可以显著改善ALK突变肿瘤患者的生存期和生活质量。
然而,长期使用一代ALK抑制剂可能会出现耐药现象,这是由于ALK基因突变引起的。
此外,一代ALK抑制剂的毒副作用也较为明显,包括胃肠道反应、血管性水肿、心律失常等。
因此,为了进一步提高疗效和减轻副作用,研究人员开始研发二代ALK抑制剂。
二代ALK抑制剂是在一代ALK抑制剂的基础上进一步改良和优化的药物。
二代ALK抑制剂包括Brigatinib(布地奈胺)、Lorlatinib(洛拉替尼)等。
二代ALK抑制剂在结构上与一代药物相似,但具有更强的抑制活性和更广的耐药性谱。
相比于一代ALK抑制剂,二代药物更能抑制包括ALK突变株和获得性耐药株在内的多种突变体。
二代ALK抑制剂不仅能够更有效地控制肿瘤生长和扩散,而且在耐药性方面也表现更出色。
此外,二代药物的毒副作用也相对较轻,例如胃肠道反应和心律失常的发生率比一代药物低。
这使得二代ALK抑制剂成为目前治疗ALK突变肿瘤的首选药物。
在划分ALK抑制剂一代和二代产品时,主要考虑以下几个方面的因素:一是药物的研发时间和上市时间,二是药物的治疗效果和耐药性谱,三是药物的副作用和安全性。
一代ALK抑制剂是最早研发的针对该靶点肿瘤的药物,并且在临床上证明了一定的治疗效果。

不可不知的靶向药1.抗EGFR靶向药厄洛替尼Tasceva【商品名】:特罗凯【药理机制】:EGFR TKI(酪氨酸激酶抑制剂,tyrosine kinase inhibitors)抑制剂【适用症】:可试用于两个或两个以上化疗方案失败的局部晚期或转移的非小细胞肺癌的三线治疗。
本适应症是基于前述国外一项Ⅲ期临床研究结果得出。
对于中国人非小细胞肺癌二线治疗的疗效尚待进一步临床研究证实。
两个多中心安慰剂对照随机的Ⅲ期试验中一线治疗局部晚期或转移性NSCLC患者,结果显示在铂类为基础的化疗(卡铂+紫杉醇;或者吉西他滨+顺铂)同时服用厄洛替尼无临床受益,因此不推荐用于上述情况的一线治疗。
【常见不良反应】:皮疹、腹泻、食欲减退、疲乏、呼吸困难、恶心呕吐、口腔炎、结膜炎、角膜结膜炎、间质性肺炎等。
【规格】:150mg每片,每盒7片。
吉非替尼Iressa【商品名】:易瑞沙【药理机制】:EGFR TKI(酪氨酸激酶抑制剂,tyrosine kinase inhibitors)抑制剂【适用症】:适用于治疗既往接受过化学治疗的局部晚期或转移性非小细胞肺癌(NSCLC)。
既往化学治疗主要是指铂剂和多西紫杉醇治疗。
本品对于既往接受过化学治疗的局部晚期或转移性非小细胞肺癌患者的疗效,是基于大规模安慰剂对照临床试验预设亚洲亚组的生存优势(注该试验总体人群中未显示改善疾病相关症状和延长生存期)及中国非对照临床试验的生存数据而确立的。
两个大型的随机对照临床试验结果表明:吉非替尼联合含铂化疗方案一线治疗局部晚期或转移性NSCLC未显示出临床获益,所以不推荐此类联合方案。
【常见不良反应】:皮疹、腹泻、皮肤溃烂、肝功能异常、出血、呼吸困难、间质性肺炎等。
【规格】:250mg每片,每盒10片。
2.抗ALK靶向药克唑替尼crizotinib【商品名】:赛可瑞【药理机制】:竞争性ATP抑制剂,主要作用靶点为ALK、c-Met、ROS【适用症】:用于治疗间变性淋巴瘤激酶(ALK)阳性的局部晚期和转移的非小细胞肺癌(NSCLC)【常见不良反应】:视力障碍、恶心、呕吐、腹泻、水肿、便秘、谷丙转氨酶升高、嗜中性白细胞减少等。

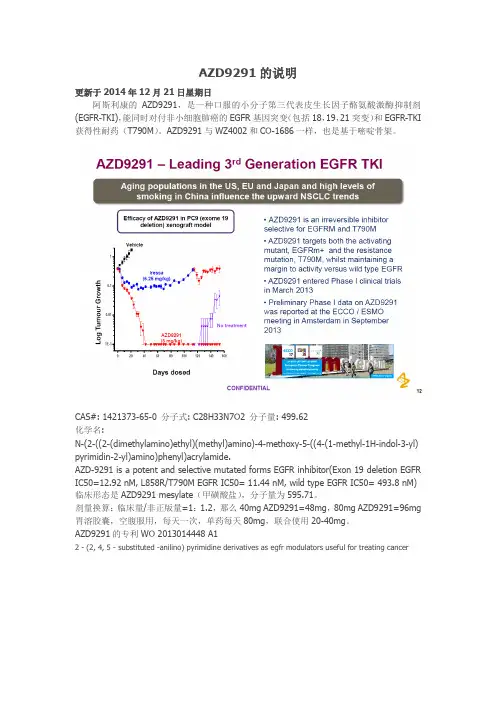
AZD9291的说明更新于2014年12月21日星期日阿斯利康的AZD9291,是一种口服的小分子第三代表皮生长因子酪氨酸激酶抑制剂(EGFR-TKI),能同时对付非小细胞肺癌的EGFR基因突变(包括18,19,21突变)和EGFR-TKI 获得性耐药(T790M)。
AZD9291与WZ4002和CO-1686一样,也是基于嘧啶骨架。
CAS#: 1421373-65-0 分子式: C28H33N7O2 分子量: 499.62化学名:N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl) pyrimidin-2-yl)amino)phenyl)acrylamide.AZD-9291 is a potent and selective mutated forms EGFR inhibitor(Exon 19 deletion EGFR IC50=12.92 nM, L858R/T790M EGFR IC50= 11.44 nM, wild type EGFR IC50= 493.8 nM) 临床形态是AZD9291 mesylate(甲磺酸盐),分子量为595.71。
剂量换算:临床量/非正版量=1:1.2,那么40mg AZD9291=48mg,80mg AZD9291=96mg 胃溶胶囊,空腹服用,每天一次,单药每天80mg,联合使用20-40mg。
AZD9291的专利WO 2013014448 A12 - (2, 4, 5 - substituted -anilino) pyrimidine derivatives as egfr modulators useful for treating cancer体外肿瘤细胞试验显示,AZD9291的耐药时间比易瑞沙、阿法替尼和WZ4002更久。

egfr-tki发展历程EGFR-TKI发展历程可以追溯到20世纪90年代末。
EGFR(表皮生长因子受体)是一种膜上酪氨酸激酶受体,被广泛表达在多种细胞类型中。
EGFR激活后可以促进细胞增殖、分化和迁移,并与肿瘤的生长和进展密切相关。
最早的EGFR-TKI是20世纪90年代末开发出来的。
它们通过与EGFR的ATP结合位点竞争性抑制其酪氨酸激酶活性,从而抑制肿瘤细胞的增殖。
最早的EGFR-TKI是Iressa(吉非替尼),于2003年获得美国食品药物管理局(FDA)批准用于非小细胞肺癌(NSCLC)的治疗。
然而,随着临床研究的进行,发现吉非替尼在治疗中的耐药性问题。
为了克服耐药性问题,新一代的EGFR-TKI被开发出来。
其中一种是Tarceva(他替尼),于2004年获得FDA批准用于NSCLC的治疗。
与吉非替尼相比,他替尼具有更强的抑制作用,并能延长患者的生存期。
然而,他替尼也存在耐药性问题,使得科研人员继续努力寻找更有效的药物。
随后的几年里,新一代的EGFR-TKI陆续开发出来。
其中包括Icotinib(易瑞沙,2011年获批)、Gefitinib(吉非替尼,2003年获批)、Afatinib(阿法替尼,2013年获批)等。
这些药物通过不同的机制抑制EGFR激酶活性,并在临床上显示出更好的疗效和耐药性管理。
近年来,EGFR-TKI的发展也在不断进行中,例如第三代EGFR-TKI Osimertinib(奥西替尼,2015年获批)已取得显著的疗效,并成为一线治疗的选择。
此外,还有许多新的EGFR-TKI正在研发中,旨在进一步提高疗效和减少耐药性。
总体而言,EGFR-TKI作为一类靶向治疗肿瘤的药物,经过多年的发展和进步,已经成为肺癌和其他EGFR突变相关肿瘤的重要治疗手段之一。
未来,随着科学技术的进步和理解的深入,有望开发出更有效的EGFR-TKI,为肿瘤患者带来更好的生存和生活质量。
JAK/STAT
JAK/STAT(Janus激酶/信号转导子和转录激活子)信号通路将来自细胞外的化学信号传递给细胞核,导致与免疫、增殖、分化、凋亡和肿瘤发生等相关基因的DNA转录和表达。
此信号通路是众多细胞因子信号转导的共同途径,其活性在炎性疾病和血液恶性肿瘤等疾病治疗研究中具有重要意义。
JAK-STAT信号级联由三个主要成分组成:由酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT三个成分组成。
JAK/STAT通路转导过程
细胞因子,如干扰素、白细胞介素和生长因子等配体,与细胞表面受体结合,引起受体分子二聚化。
与受体偶联的JAK 相互接近并通过交互的酪氨酸磷酸化而被激活。
活化后的JAK使受体的酪氨酸磷酸化,为具有SH2结构域的STAT创建结合位点。
STAT结合至受体后,在JAK作用下,STAT酪氨酸705(Tyr 705)被磷酸化。
STAT就会从受体上脱离,形成同/异二聚体。
STAT二聚体进入细胞核后,会结合特定的调节序列以激活或抑制靶基因的转录。
JAK/STAT信号通路图
按靶点分类:。


基于质谱的蛋白质激酶-小分子相互作用研究进展陈津;王方军【摘要】蛋白质激酶在生命过程中起着关键的调节作用,蛋白质激酶的异常往往会导致恶性疾病如癌症的发生.靶向蛋白质激酶的小分子抑制剂为相关疾病的治疗提供了切实可行的方案,因此研究蛋白质激酶-小分子的相互作用对于靶向药物的开发具有重要作用.质谱作为一种高精度、高通量、高灵敏度的分析仪器,在蛋白质-小分子相互作用研究领域的应用得到了快速发展.本综述总结了质谱技术应用于蛋白质激酶-小分子相互作用研究的主要策略和最新进展,探讨比较各种方法的优缺点.基于质谱的分析方法将有助于从分子水平上深刻揭示蛋白质激酶-小分子的相互作用机理,为靶向药物的设计开发提供新的思路.【期刊名称】《世界科学技术-中医药现代化》【年(卷),期】2018(020)008【总页数】8页(P1314-1321)【关键词】蛋白质激酶;小分子抑制剂;相互作用;激酶;质谱【作者】陈津;王方军【作者单位】福建厘科大学临床分子诊治研发中心,福建医科大学附属第二医院泉州362000;中国科学院分离分析化学重点实验室,中国科学院大连化学物理研究所大连116023;中国科学院分离分析化学重点实验室,中国科学院大连化学物理研究所大连116023【正文语种】中文【中图分类】R311正常细胞中蛋白质激酶受到严格调控,蛋白质激酶表达量和活性发生变化将引起细胞生物学过程的紊乱甚至导致癌症等恶性疾病的发生。
早在20世纪70年代末期,科研人员就发现了激酶和癌症的联系,激酶活性不受控制将会引起细胞性状发生改变最终导致疾病[1-3]。
利用靶向小分子抑制剂或激动剂对特定蛋白质激酶的活性进行特异性调控能为相关疾病提供有效的治疗方案,小分子抑制剂也成为了目前最重要的抗肿瘤药物之一。
截止2017年,共有38种小分子激酶抑制剂类药物获美国FDA批准并应用于临床。
目前已经有5000多种蛋白质激酶和蛋白质激酶-抑制剂复合体的晶体结构被成功解析,极大促进了人们对抑制剂作用机理的理解并为新型小分子抑制剂药物的设计合成提供了重要参考。
VEGF及其抑制剂在肿瘤治疗中的研究进展[摘要]内皮血管内皮生长因子(vacularendothelialgrowthfactor,VEGF)是一种二聚体糖蛋白,能够刺激内皮细胞的增殖、生存与迁移,在多种肿瘤细胞中均有较高水平的表达。
其不仅是肿瘤血管生成过程中最重要的促血管生成因子之一,在肿瘤免疫中也发挥了重要的作用。
目前,已有不同类型的VEGF抑制剂被投入临床,用于肿瘤的治疗。
本文针对VEGF 家族成员、VEGF在肿瘤发生发展中的活性及VEGF相关抑制剂的研究进展进行综述。
[关键词]肿瘤;VEGF;血管生成;肿瘤免疫;抑制剂血管生成指已有血管通过出芽或套叠的方式形成新血管的过程,在胚胎发育、伤口愈合等生理过程或糖尿病性视网膜病、肿瘤等病理过程中都发挥着重要的作用。
血管生成过程复杂,有许多调控因子参与。
其中,血管内皮生长因子(vacularendothelialgrowthfactor,VEGF)家族及其受体(VEGFR),是促进血管细胞分化和血管形成最重要的组织因子。
本文主要介绍了VEGF及其受体家族成员、VEGF在肿瘤中的生物活性以及VEGF 抑制剂在肿瘤治疗方面的研究进展。
1、VEGF家族成员及其受体VEGF最初在牛垂体分泌物中被发现,直至1989年,Abraham等人将其分离纯化,并命名为血管生成因子。
目前该蛋白家族共发现VEGFA、VEGFB、VEGFC、VEGFD、VEGFE(病毒编码)以及胎盘生长因子(placentalgrowthfactor,P1GF)几种常见异构体,两种不VEGFl45、VEGFl83常见异构体,以及两种抑制性异构体VEGFl65b、VEGF-A,但其抑制机制依然存在争议。
VEGFA是一种45KD的同型二聚体糖蛋白,是该家族成员中最具特异性、功能性最强的血管生成因子,即我们通常所说的VEGF。
VEGF受体家族有VEGFR-1、VEGFR-2、VEGFR-3三个成员。
肿瘤小分子靶向药物分类肿瘤小分子靶向药物分类如今肿瘤的治疗手段多元化,其中靶向治疗为较新兴的治疗方式,由于毒副作用较小,疗效较突出,使得靶向治疗的成本也相对高昂。
分子靶向药物是在分子生物学、分子遗传学理论基础上出现的新药, 因其精确的靶向治疗作用,相对于传统化疗药物有很多优势, 形成了一门治疗肿瘤的新领域,为肿瘤的治疗提供了一种不良反应较小的方法。
近20年来,随着医学科学的发展,大量以肿瘤细胞水平表达为靶点的新的抗肿瘤药物不断问世,并逐渐走向临床, 主要包括细胞信号转导分子抑制剂、新生血管抑制剂、靶向端粒酶抑制剂以及针对肿瘤耐药的逆转剂。
攻击肿瘤的靶点有多方面, 目前研究较成熟的主要有肿瘤细胞表面的靶点 (抗原或抗体 ), 如细胞膜分化相关抗原 (CD13,CD20,CD22,CD33,CD52,CD117等) , 细胞信号转导分子如表皮生长因子(EGF)及其受体(EGFR)和血管内皮生长因子(VEGF)及其受体上的酪氨酸激酶, 以及法尼基转移酶, 基质金属蛋白酶等。
分子靶向药物目前尚无统一的分类方法。
根据作用靶点不同,可分为以下4类。
●蛋白激酶细胞的分化信号传导因子中, 含有大量的蛋白激酶家族。
在细胞信号传导过程中, 蛋白酪氨酸激酶十分重要, 它可催化ATP上的磷酸基转移到许多重要蛋白质酪氨酸残基上使其磷酸化, 导致传导支路的活化, 影响细胞生长、增殖和分化, 而许多肿瘤细胞中酪氨酸激酶活性异常升高。
超过50%的癌基因及其产物具有蛋白酪氨酸激酶活性, 它们的异常表达将导致肿瘤的发生。
此外, 该酶的异常表达还与肿瘤转移、肿瘤新生血管生成、肿瘤对化疗耐药有关。
研究能阻断或修饰由信号传导失常引起疾病的选择性蛋白激酶抑制剂, 被认为是有希望的药物开发途径。
目前, 已经发现了一些蛋白激酶抑制剂和针对不同蛋白激酶 ATP结合位点的小分子治疗剂, 并已进入临床研究,如酪氨酸激酶抑制剂吉非替尼、厄洛替尼等及法尼基转移酶抑制剂安卓健等。
MAPK丝裂原活化蛋白激酶(MAPK)信号通路介导信号从细胞表面向细胞核内转导,通过三级激酶级联的形式传导细胞外信号,调控着细胞的生长、分化、炎症、凋亡、癌化、肿瘤细胞的侵袭和转移等多种生理活动过程。
MAPK通路参与了许多疾病的发展,包括阿尔茨海默病(AD)、帕金森病(PD)、肌萎缩性侧索硬化(ALS),在癌症、免疫及神经退行性疾病的治疗中发挥了重大作用。
MAPK通路转导过程MAPK家族在哺乳动物细胞中3个经典转导通路:MAPK(ERK)、C-Jun N末端kinse/应激激活蛋白激酶(JNK/SAPK)和p38激酶。
每个与MAPK相关的级联反应由不少于三种酶串联激活:MAPK激酶激酶(MAPKK)、MAPK 激酶(MAPKK)和MAPK激酶(MAPK)。
MAPK通路被多种细胞外和细胞内刺激激活,包括肽生长因子、细胞因子、激素和各种细胞应激源。
在ERK信号通路中,ERK1/2被MEK1/2激活,而MEK1/2被Raf激活。
Raf被Ras-GTPase激活,其激活是由表皮生长因子受体等RTKs诱导的。
JNK和p38 MAPK信号通路被不同类型的细胞应激激活。
JNK路径由JNK(一种MAP2K(如MKK4(SEK1)或MKK7))和MAP3K(如ASK1、TAK1、MEKK1或MLK3)组成。
在p38通路中,p38被MKK3或MKK6激活,这些MAP2K被JNK通路中功能相同的MAP3K激活。
MAPK信号通路图Selumetinib (AZD6244) 606143-52-6 MEK1 14 nM *RafAZ 628 878739-06-1 无细胞BRAF,BRAFV600E105 nM *p38 MAPK*JNK。
FDA批准的激酶小分子抑制剂类药物及分类一览
蛋白激酶
蛋白激酶(Kinase)是细胞生命活动重要的信号使者,可催化将ATP末端的γ-磷酸基团转移至底物上,从而将各种信号进行传递(图1)。
蛋白激酶参与了众多的生理过程,包括细胞增殖、存活、凋亡、代谢、转录以及分化等。
药理学及病理学研究表明,对于很多疾病,如肿瘤、炎症性疾病、中枢神经系统疾病、心血管疾病及糖尿病等,蛋白激酶都是一个理想的药物靶点。
图1 Mechanism of protein kinases and related publications
对于蛋白激酶的研究始于20世纪50年代,并在90年代随着MAPK/ERK、JAK 及PI3K等信号通路的揭示而达到一个研究热潮。
迄今为止,在人体中发现了518种蛋白激酶,而编码具有激酶活性蛋白的基因则高达900多种。
与之相对应,有关激酶抑制剂的研究也逐步发展,并在激酶作用机制的阐明过程中扮演了
重要角色,并成为重要的药物研究热点。
该领域研究的文献数量也是逐年上升,从侧面反映了其在基础研究和药物发现中的重要性。
蛋白激酶抑制剂及其分类
过去的15年间,激酶抑制剂作为药物候选的研究取得了长足的进步,不论是基础研究还是在工业界。
在人体现有药物靶点里面,蛋白激酶家族成员占比高达10%(FDA批准药物分子靶点深度解读)。
2001年,第一个激酶抑制剂类药物Imatinib获得FDA批准,成为该领域发展的里程碑,此后十年该类药物以平均每年获批一种的速度稳步发展。
而在2012年1月至2015年2月期间,小分子激酶抑制剂类药物迎来爆发式发展,共有15种新药获得审批。
截至2016年12月底,共有31种小分子激酶抑制剂类药物获得审批,同时还有大量的化合物处于临床或临床前研究中。
除此之外,科研人员还解析了超过5000种的蛋白激酶或蛋白激酶-抑制剂复合体的晶体结构,且超过五分之一的人类蛋白激酶具有明确的小分子抑制剂。
因此,小分子激酶抑制剂已成为药物研发的一个热点领域。
蛋白激酶尽管在一级序列上有所差异,但在三维结构上却具有高度的保守性,特别是在催化活性结构域附近。
该区域存在一个β-折叠构成的N-lobe区域及α-螺旋构成的C-lobe区域,而ATP结合在两者构成的沟状区,也是很多激酶抑制剂的结合位点。
活性位点附近还存在一条Activation-Loop,通常末端存在一个保守的Asp-Phe-Gly (DFG)结构基序(图2A)。
图2 Kinase structure and different types of reversible small-molecule kinase inhibitor
根据结合模式的不同,激酶抑制剂可分为不可逆及可逆两大类。
前者指化合物通过与Cys反应形成共价键结合在ATP结合位点上,从而封闭ATP的结合空间,该过程具有不可逆转性。
后者根据结合口袋区域及DFG基序构象的不同,可分为四种主要的不同亚型(图2B)。
类型Ⅰ为ATP竞争性抑制剂,结合与活性形式激酶DFG基序上的Asp残基。
类型Ⅱ抑制剂结合与非活性状态的激酶中,其上DFG基序上的Asp残基朝向分子外侧。
而类型Ⅲ的抑制剂结合在ATP附近别构
位点上,但同ATP结合口袋没有相互作用。
类型Ⅳ抑制剂结合在远离ATP结合
位点的别构作用区域。
还有些激酶抑制剂,如双底物类抑制剂,可以统一划分在类型Ⅴ里面,作用模式与上述四类有所不同。
图3 Small molecular kinase inhibitors approved by FDA in 2001-2016
在获批的31中小分子激酶抑制剂类药物中(图3),绝大多数为酪氨酸激酶抑制剂,还有些属于丝氨酸/苏氨酸激酶抑制剂,只有2014年七月获批的Idelalisib 属于脂激酶类抑制剂。
根据靶点蛋白及药物属性的不同,分类整理如下:
1. FDA批准的可逆NRTK(非受体型酪氨酸激酶)抑制剂类药物
图4 Reversible NITK inhibitors
2. FDA批准的可逆RTK(受体型酪氨酸激酶)抑制剂类药物
图5 Reversible RTK inhibitors-1
图6 Reversible RTK inhibitors-2
3. FDA批准的不可逆蛋白激酶抑制剂类药物
图7 Inreversible protein kinase inhibitor
4. FDA批准的丝氨酸/苏氨酸激酶抑制剂类药物
图8 Serine/Threonine kinase inhibitors
5. FDA批准的脂激酶抑制剂类药物
图9 Lipid kinase inhibitor
挑战与展望
在过去的15年里,基于蛋白激酶的药物发现取得了巨大进步。
相比于GPCR,膜通道与转运蛋白及蛋白酶等传统药物靶点领域,激酶抑制剂代表了一类年轻而又充满活力的药物发现空间,并取代GPCR成为癌症领域最火热的细胞治疗靶点。
尽管在短短十五年时期内已有31种药物获得批准,但在2016年,近五年的快速增长势头没能继续延续。
激酶小分子抑制剂类药物研究领域仍面临诸多挑战。
首先,人体内激酶家族中只有少数成员获得了详尽研究。
绝大多数抑制剂研究集中于酪氨酸激酶及类酪氨酸激酶家族,如CDK、MAPK、GSK3、CMGC、PKA、PKG及PKC等。
这一不平衡还体现在现有获批药物数量上,针对BCR-Abl、ErbBs 及VEGFRs三类酪氨酸激酶开发的药物占到了总数的三分之二以上。
而在另一分类体系中,脂激酶只有一种抑制剂类药物上市,尽管这类酶的抑制剂早在20世纪90年代就早已有报道,且开展了很多临床及临床前研究。
以上现状表明,人体激酶组中的很多成员还处于被冷落状态。
我们需要开发出新型的研究方法或筛选探针,以便更好的揭示这些激酶的作用机制,并有效开发其作为药物靶点的潜力。
令人鼓舞的是,最近几年有数个激酶成员的抑制剂类药物首次获得批准,如2013年MEK抑制剂Trametinib、BTK的抑制剂Ibrutinib及2015年获批的CDK抑制剂Palbociclib。
其次,尽管激酶的梯级调控信号调节着众多的生理过程,如炎症反应、中枢神经系统异常、心血管疾病、糖尿病及癌症等,但现有获批药物超过90%以上集中在癌症治疗领域。
针对激酶的小分子抑制剂类药物应用领域亟待拓展。
目前,Tofacitinib已被批准用于关节炎治疗。
即便是在肿瘤治疗领域,也有很多问题有待进一步深入研究,如肿瘤的耐药性问题以及同其他药物的联合用药等。
第三,现有的很多激酶抑制剂药物存在较多相似性,大多是在已批准药物基础上设计优化而成,如针对ErbB的五种抑制剂均包含相同的核心结构模块。
从这个角度讲,现有药物的结构多样性不够丰富,亟需增加药物研发初始阶段高通量筛选(HTS)所用化合物库的结构多样性。
而天然产物库,通常包含不同于绝大多数
化学合成抑制剂库的药效团及分子骨架,有望成为拓展HTS库结构多样性的新资源。
第四,具有新的作用机制及特异性的激酶抑制剂类药物有待开发。
由于激酶ATP 结合口袋区域附近结构的保守型,绝大多数抑制剂为可逆性抑制剂。
这也导致了绝大多数小分子抑制剂存在不止一个作用靶点分子,导致脱靶现象以及副作用的产生。
与之相随,具有绝对选择性的抑制剂分子极为稀少。
未来工作中,我们需要探索具有新型作用机制的抑制剂分子。
而激酶抑制剂的选择性则是一个略带争议的特性。
早期研究中,具有交叉抑制活性或广谱选择性的抑制剂是肿瘤学研究的得力方法。
而当前,选择性抑制剂更适合于肿瘤治疗的理论逐步被广泛认同。
研究结论表明,激酶抑制剂类药物并不需要绝对的选择性,而是要有适当的选择性以便在药效与毒性间达到某个平衡点。
本文所引用资料:
1. Small Molecule Kinase Inhibitors as Anti-Cancer Therapeutics. Mini-Reviews in Medicinal Chemistry, 2012, 12, 399-411
2. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nature Reviews Clinical Oncology, 2016,13,209–227
3. Tyrosine Kinase Inhibitors: Views of Selectivity, Sensitivity and Clinical Performance.
2013,53,161-185
4. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016Jan;21(1):5-10
5. FDA-approved small-molecule kinase inhibitors. Trends in Pharmacological Sciences 2015,36,422-439
6. /Drugs/default.htm
7. /scripts/cder/daf/index.cfm
8. 部分药物信息查询自药渡网-药物数据。