金属有机第六章氢甲酰化反应
- 格式:pdf
- 大小:448.43 KB
- 文档页数:9




烯烃氢甲酰化技术工业化现状及展望烯烃氢甲酰化是一种重要的有机合成方法,可以将烯烃和一氧化碳在催化剂的作用下,转化为对应的甲酰化合物。
该技术具有丰富的原料资源、高效、环境友好等优势,因此在有机合成领域有着广泛的应用前景。
本文将就烯烃氢甲酰化技术的工业化现状及展望进行讨论。
目前,烯烃氢甲酰化技术在工业化中已经取得了一系列显著的进展。
特别是在催化剂的研究方面,钯催化剂是应用最广泛的一种。
通过优化催化剂的配方和制备工艺,传统的钯系催化剂具有了更高的活性和选择性。
同时,一些新型的催化剂也被开发出来,例如钯/镍合金催化剂和钯基非贵金属催化剂等,它们不仅具有较高的活性和选择性,还具有更低的成本。
与此同时,催化剂的载体及表面修饰等方法也被用于优化催化剂的性能。
另外,反应条件的优化也对烯烃氢甲酰化技术的工业化起着重要作用。
常见的反应条件包括反应温度、压力、气体流量、溶剂种类等。
在反应温度方面,一般而言,较高的温度有利于反应的进行,但同时也会导致产物的选择性变低。
因此,在工业化应用中,需要平衡活性和选择性之间的关系。
此外,溶剂的选择也至关重要,一方面可以提高反应速率,另一方面也可以改善产物的分离纯化。
因此,对于溶剂种类的研究和优化也是烯烃氢甲酰化技术工业化的关键之一。
展望未来,烯烃氢甲酰化技术的工业化仍然面临一些挑战和机遇。
首先,活性和选择性仍然是催化剂研究的重点。
虽然目前的催化剂已经具备了一定的活性和选择性,但仍需进一步提高。
对于新型催化剂的研究和发现,以及对催化机理的深入理解,有助于优化催化剂的性能。
反应条件的优化仍然值得关注。
通过改进反应温度、压力、气体流量等参数,可以更加精确地控制反应过程,提高产物的选择性和收率。
此外,对于反应过程中产生的副产物的处理也需要深入探讨和优化。
进一步研究烯烃氢甲酰化技术在可持续发展方面的应用也是未来的方向之一。
如何降低反应的碳足迹,减少对环境的影响,是当前研究的重要课题。
可以通过开发新型催化剂、提高反应条件的效率等方式来实现可持续发展的目标。




合肥市第一中学2024-2025学年高三上学期期中教学质量检测化学学科试卷时长:75分钟分值:100分可能用到的相对原子质量:H -1C -12O -16Na -23Mg -24S -32Ca -40Ce -140第Ⅰ卷(选择题)一、单选题:本大题共14小题,每小题3分,共42分。
1.在巴黎奥运会舞台上,科技与体育双向奔赴,释放更加迷人的魅力。
下列说法不正确的是( )A.巴黎奥运会火炬的燃料主要成分丙烷属于烃类B.中国队领奖服采用的是环保再生纤维材料,该材料为有机高分子材料C.乒乓球台可变灯光系统的控制芯片的主要成分是硅D.生产弹性地板的原材料天然橡胶属于烃的衍生物2.下列化学用语表述不正确的是( )A.次氯酸钠的电子式:B.固体中的链状结构:C.铜的基态原子的简化电子排布式:D.的VSEPR 模型3.物质的微观结构决定宏观性质,进而影响用途。
下列结构或性质不能解释其用途的是( )选项结构或性质用途A高铁酸钠具有强氧化性高铁酸钠可用于水体的消毒B聚乳酸中含有酯基聚乳酸可用于制备降解材料C 冠醚18—冠—6空腔直径(260~320pm )与直径(276pm )接近冠醚18—冠—6可识别,能增大在有机溶剂中的溶解度D二氧化硫具有一定的氧化性二氧化硫可用作漂白剂4.化学是一门以实验为基础的学科。
下列图示能达到实验目的的是()HF []92Ar 3d 4s23SO K +K +4KMnOA.图1可用于熔化固体烧碱B.图2中的装置可实现反应随关随停C.利用图3装置通过蒸干溶液制备晶体D.利用图4装置模拟工业制备并检验5.化合物是一种用于合成药物的重要试剂,其结构简式如下图所示。
已知、、、、为原子序数依次增大的前四周期主族元素,、位于同一主族,、、的最外层电子数之和等于的最外层电子数。
下列有关叙述错误的是()A.简单离子半径:B.简单氢化物稳定性:C.化合物与均为共价化合物D.化合物中所有原子均满足8电子相对稳定结构6.目前,世界上金属镁的主要生产方法有以下两种:方法1:皮江法:方法2:无水氯化镁熔盐电解法:下列有关叙述错误的是()A.采用皮江法制镁能耗高,且不利于促进碳中和B.采用皮江法制得金属镁,至少排放C.可用代替还原(沸点:、)D.直接加热可获得电解法制镁的原料无水7.一种微生物一光电化学复合系统可高效实现固定并生成,其原理如图所示。
瓦克法(Wacker process),又称Hoechst-Wacker法,最早是指乙烯在含有四氯钯酸盐催化剂的水中,被空气中的氧气氧化为乙醛的反应。
[1][2][3][4][5][6]这是第一个工业化的有机金属(有机钯)反应,亦是均相催化和配位催化中很重要的一个反应,在1960年代后发展很快,在石油化工发达的国家已大幅取代了乙炔水合法,用于从烯烃制取醛、酮类。
反应中的钯配合物与烯烃配合物蔡氏盐类似,不过后者是一个异相催化剂。
此反应形式上与氢甲酰化反应类似,都是工业上用于醛类的反应。
但两者不同的是,氢甲酰化所用的是铑基催化剂,而且氢甲酰化是一个增碳过程。
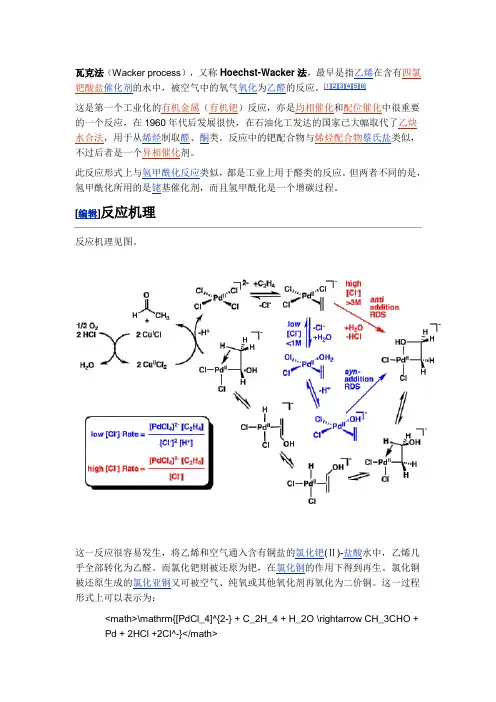
[编辑]反应机理反应机理见图。
这一反应很容易发生,将乙烯和空气通入含有铜盐的氯化钯(Ⅱ)-盐酸水中,乙烯几乎全部转化为乙醛。
而氯化钯则被还原为钯,在氯化铜的作用下得到再生。
氯化铜被还原生成的氯化亚铜又可被空气、纯氧或其他氧化剂再氧化为二价铜。
这一过程形式上可以表示为:<math>\mathrm{[PdCl_4]^{2-} + C_2H_4 + H_2O \rightarrow CH_3CHO + Pd + 2HCl +2Cl^-}</math><math>\mathrm{Pd + 2CuCl_2 + 2Cl^- \rightarrow [PdCl_4]^{2-} +2CuCl}</math><math>\mathrm{2CuCl + 1/2O_2 + 2HCl \rightarrow 2CuCl_2 +H_2O}</math>[编辑]Wacker-Tsuji氧化反应为氧化更复杂的底物,Tsuji 发展了混合溶剂体系(DMF/H2O),又称为“Wacker-Tsuji 氧化反应”。
最常用的烯烃底物是末烯,反应遵循马氏规则,得甲基酮。
1,2-二取代烯烃也可反应,但区域选择性难以控制。

基础化学无机部分1章习题1.举例说明什么是拉平效应?为什么水不能区分H2SO4, HNO3, HClO4的酸性强弱,也不能区分O2-, NH2-, Ph3C-的碱性强弱?2.BrF3自电离产物是什么?用VSEPR写出BrF3和自电离产物的几何构型。
3.对非水液氨、醋酸和硫酸,写出:4.它们各自的自电离方程。
5.把CH3COOH分别置于上述三种溶剂中,各发生什么变化?写出反应方程式。
6.把b的三种溶液和纯溶剂相比,原溶剂的酸碱性各有何变化?溶质在各溶剂中是酸还是碱?7.为什么在强酸性溶剂中制备阳离子物种如I2+和Se82+,而在强碱性溶剂中制备阴离子物种如S42-, Pb94-?8.亚磷酸H3PO3的结构式可表示为P(OH)3和HPO(OH)2两种异构体,实验测得亚磷酸的pKa值为1.8,判断亚磷酸的结构应该是什么?次磷酸的pKa为2.00,写出其正确的结构式。
9.把NR3, S2-, NF3, O2-, NH3, OH-, NCl3, N3-按照碱性大小排序,并解释。
10.BH3和BF3都可和(CH3)2NPF2生成加合物,在一种加合物中,B原子和N相连,另一种则和P相连,绘出两种结构式,并说明形成两种结构的原因。
2章习题11.2价Ni配合物[Ni(PPh3)2Cl2]为顺磁性,Pt的类似配合物[Pt (PPh3)2Cl2]为反磁性,写出每种化学式的所有异构体。
12.讨论下两组异构体的偶极矩偶极矩情况:(a) 顺式和反式的MA2B4,(b)经式和面式的MA3B3。
13.绘出[Co(en)2Cl2]+、[Co(en)2NH3Cl]2+和[Co(en)(NH3)2Cl2]+的所有几何和光学异构体。
14.[Co(en)3 ]3+、[Ru(bpy)3]2+和[PtCl(dien)]+ (dien二乙三胺)中,哪些是手性化合物?15.Mo(CN)84-水溶液作13C的NMR表征,只得到一个峰,由此你得到什么结构信息?16.绘出五氨络钴(III)-μ-硫氰根-五氰络钴(III)的结构。

教学目标:了解烯烃羰基化反应
教学重点:了解工业上合成羰基化合物的方法
教学安排:F
> F7;10min
1—
一、羰基化反应
在烯烃分子中引进羰基的反应称为羰基化反应,又称羰基合成反应,它是一类反应的总称。
使用不同原料,产物不同,但都生成碳基衍生物,这是20世纪40年代发展起来的烯烃络合催化反应,在有机化工上有重大应用。
在上述反应中,相当于烯烃的双键与甲酰基衍生物加成反应,如反应(1)相当于烯烃双键的一端引入H,另一端引入甲酰基,故称为烯烃氢甲酰化反应:
在反应(2)~(6)中相当于烯烃双键的一端引入H,另一端分别引入-COOH、-COOR'、-CONR'2、-CO-OOCR和-COCl,生成羧酸、酯、酰胺、酸酐和酰氯,因此分别称为氢羧化、氢酯化、氢酰胺化、氢酐化和氢酰氯化反应。
以烯烃的氢甲酰化反应为例,说明这类反应的特点。
1.在形式上,H-CHO的氢可以加到双键的任一个碳上,形成正构醛和异构醛:
2.两种产物的比例取决于烯烃的结构,使用的催化剂和反应条件。
3.内烯烃进行氢甲酰化反应,在反应过程中双键自动异构到分子链端上,也是形成正构醛和异构
醛。
4.使用有手性配体的催化剂,可进行不对称合成。
工业上利用这种方法生产大吨位的醛。
改变催化剂和CO/H2进料比例,可以一步得到醇。
其它类型的羰基化反应也应于工业生产,如乙烯进行氢羧化反应是合成丙酸的工业方法:
二、关键词
羰基化反应,羰基合成反应,氢甲酰化,氢羧化,氢酯化。
氢酰胺化,氢酐化,氢酰氯化,手性配体,不对称合成。
膦配合物在合成中的若干应用摘要:近年来,合成手性配体的一种发展趋势是,在手性膦配体的基础上,引入氮、氧或硫等杂原子,生成多齿的混合功能团配体。
本文选了几个方面来综述膦配合物的应用。
关键字:膦配合物;氢甲酰化;Suzuki偶联反应1.引言手性化合物在医药、农药、食品添加剂、功能高分子材料等领域有着广泛的应用。
对于许多有生物活性的化合物而言,其两种对映体往往具有不同程度的活性,或具有完全不同的生理作用。
因此如何能够高产率、高光学纯度、低成本低能耗地获得单一对映体化合物是化学家研究的重要课题。
研究表明,不对称催化是制备单一对映体化合物最有效、最重要的途径之一。
它仅用少量手性催化剂,实现手性转移、手性倍增,生成大量的旋光性产物。
为实现高效的不对称催化反应, 化学工作者需要设计合成优秀的手性催化剂。
通常,手性金属催化剂由金属中心和中性或阴离子的手性辅助配体组成。
借助手性配体的结构、构型和刚性,能够在催化活性中心周围营造一种特殊的手性立体微环境,以有效控制反应物分子的空间取向和反应进程,发挥特异的光学选择性。
因此手性配体是实现不对称催化反应的关键。
近年来,合成手性配体的一种发展趋势是,在手性膦配体的基础上引入氮、氧或硫等杂原子,生成多齿的混合功能团配体。
这类配体中的混合功能团,不仅能与金属中心络合,生成刚性较强的手性金属络合物,而且在催化反应的过程中,配体中功能团还能够与底物络合,生成活性中间络合物,类似于酶催化中金属中心及其周围功能团与底物分子的多点相互作用,减少了过渡态的自由度,有利于提高光学选择性。
本文选了几个方面来综述膦配合物的应用。
[9]2.氢甲酰化反应方面氢甲酰化反应是烯烃与合成气在过渡金属络合催化剂作用下生成醛或醇的均相催化反应过程。
氢甲酰化反应最早研究的催化剂是Fe(Co)6、Co2(CO)8和Rh4(CO)12等,这类催化剂需在高温高压下操作, 热稳定性差且选择性也不高。
经过一系列探索研究后壳牌公司最早提出了用有机膦配体部分地取代CO配体的改性途径,改性后的催化剂由于引入了比CO体积大且具有更强的电子给予能力的膦配体, 其热稳定性和选择性都有了显著提高,从而实现了低温低压氢甲酰化反应的操作过程。
双膦配体铑化合物用作催化剂
2016-06-09 12:54来源:内江洛伯尔材料科技有限公司作者:研发部
铑/双膦配体催化均相内烯烃氢甲酰化反应
烯烃氢甲酰化反应是指烯烃与CO和H2在催化剂的作用下生成醛的反应, 产物醛绝大部分被进一步加工成醇、羧酸、胺和羟基醛等产品. 其中醇的最大用途是用作溶剂及合成表面活性剂和增塑剂的原料. 随着塑料加工、汽车工业、电缆工业以及建筑业的发展, 对醇的需求将越来越大.
相对于末端烯烃而言, 内烯烃价廉易得, 因此内烯烃氢甲酰化反应的
研究日益受到重视. 人们不断探索和研究新的催化体系, 以期能进一步提高内烯烃氢甲酰化反应的活性和直链醛选择性. 其中对配体的改性是该研究的一个重要手段, 不同类型的配体对烯烃氢甲酰化反应的活性、选择性乃至反应动力学影响很大. 双膦配体具有特殊的电子效应和立体效应, 对铑催化烯烃氢甲酰化反应的活性和区域选择性有重要影响.
四川大学化学学院有机金属络合催化研究所陈华小组系统地综述了近年来双膦配体铑配合物催化均相内烯烃氢甲酰化反应的研究进展, 讨论了双膦配体结构对催化剂活性和生成直链醛选择性的影响.相关文章发表在《催化学报》上(题名“铑/双膦配体催化均相内烯烃氢甲酰化反应的研究进展”).。
第六章氢甲酰化反应Outline:A.Introduction and Cobalt catalystB.Cobalt Phosphine-Modified CatalystsC.Rhodium Phosphine Catalysts
D. Aqueous-Phase Rh HydroformylationE. Asymmetric Hydroformylation
Hydroformylation was discovered by Otto Roelen in 1938.
Roelen's observation that ethylene, H2and CO
were converted into propanal, and at higher pressures, diethyl ketone, marked the beginning of hydroformylation.
A. Introduction and Cobalt catalyst
Three commercial processes1. Cobalt salt2. Cobalt salt/phosphine ligand (Shell)3. Rhodium salt/phosphine ligand(Ruhrchemie/Rhone-Poulenc, Mitsubishi-Kasei, Union Carbide, and Celanese)
Both linear and branched products can be produced, although the more desirable linear aldehydesare usually the major products when using aliphatic olefins.
Hydroformylation: Thermodynamics
Thermodynamics at standard conditions:At higher temperatures the entropy loss becomes more important: ΔG will be less negative.
The TD favored product is the alkane. The obtained product is the aldehyde because of kinetic control.Mechanism of the Co-catalyzed hydroformylationAll steps are reversible, except product formation,which is often rate-determining.
Preferential formation of thelinear product is due to stericinteractions.Trapping of the alkyl by CO determines the regioselectivity.β-hydride elimination is suppressed due to the high CO pressure.
Hydrogenolysis is the rate-determining step for simple terminal olefins.
HCo(CO)4Catalyst
※The first step is a dissociative substitution of alkene for CO. ※The second step:Migratory insertion can result in either a primary or secondary metal alkyl. Although this is the step that sets the regiochemistry of the products, it is rapidly reversible. This rapid reversibility results in alkene isomerization and H/D exchange. ★Since ß-H elimination requires an open coordination site, isomerization and isotope exchange are inhibited by increased CO pressures.
※The next step ( third)is a second migratory insertion to form the coordinatively unsaturated acyl that can coordinate another CO to give the 18 electron acyl complex. Under standard catalytic conditions with 1-octene, this is the only species observed by IR.
The mechanism of the hydrogenolysis step is less clear.
An alternate bimetallic pathway was also suggested, but not favored, by Heck and Breslow.
The inverse dependence on CO pressure is consistent with the mechanistic requirement for CO dissociation from the various saturated 18e species to open up a coordination site for alkene or H2binding.
Hydroformylation: Kinetic studiesWhen using a 1:1 ratio of H2/CO, the reaction rate is essentially independent of pressure due to the opposing orders of H2and CO..Increasing the H2/CO ratio is of limited use for increasing the overall reaction rate because HCo(CO)4is only stable under certain minimum CO
partial pressures at a given temperature
Increasing the CO partial pressure decreases the
hydroformylation reaction rate and the amount of alkene isomerization side reactions, while increasing the aldehyde linear to branched product ratio.
Why? ?
Pino proposed that the apparent marked differencebetween HCo(CO)4 catalyzed hydroformylation at low and high CO partial pressures was due to the existence of two active catalyst species, HCo(CO)4and HCo(CO)3, formed
from the CO association/dissociation equilibrium:
16e-HCo(CO)318e-saturated HCo(CO)
4
Regioselectivity:
Under lower CO partial pressures an unsaturated 16e-RCo(CO)3will have a long enough lifetime to allow reverse β-hydride eliminationand increase the possibility for alkene reinsertionto the branched alkyl species, which is slightly more favored thermodynamically.The regioselectivity of HCo(CO)4(or HCo(CO)3) for producing the more valuable linear aldehydes varieswith reaction conditions and alkene substrates used.
◆High CO partial pressure slows the rate of catalysis, but increases the linear to branched aldehyde product ratio. ◆Higher CO partial pressures also lower alkene isomerization side reactions. ◆Higher temperatures increase the reaction rate, but lower the linear aldehyde product regioselectivity and increase various undesirable side reactions.
Side reactions of the product aldehydes to form heavier productsgenerally occur, particularly at higher reaction temperatures, and usually account for ~ 9% of the product distribution.
The regioselectivity can be modifiedby adding phosphineto the Co-catalyzed hydroformylation system.This advance was discovered at Shell chemical. Using HCo(CO)3PBu3as catalyst.