毛细管电泳出现问题分析
- 格式:docx
- 大小:9.58 KB
- 文档页数:5

SSR标记荧光毛细管电泳在种子检测中常见问题分析李巧英(山西省种业发展中心,太原030006)摘要:SSR分子标记荧光毛细管电泳在农作物种子质量检测中常用于种子真实性检测、种子纯度鉴定和指纹数据库的构建。
试验过程中模板DNA浓度、特异峰型的统计分析以及仪器设备运行和维护、试验结果与指纹数据库的比对等关键环节都直接影响待测样品的结果判定。
就检测中遇到的问题进行分析,并提出相应的解决方法,以期为农作物种子质量检测荧光毛细管电泳技术平台提供参考。
关键词:SSR标记;荧光毛细管电泳;种子检测;问题;分析分子标记技术是近年来发展较快的基因检测技术,随着新标记方法的开发和试验技术的改进,在基因水平上检测农作物种子信息逐渐成为监控种子质量的一个重要手段。
由于分子标记法不受自然条件和表现性状的约束,实现了种子品种遗传信息室内快速检测,受到大家的青睐。
SSR作为第2代分子标记方法,是一种共显性标记,具有重复性好、等位变异多、数据分析简便、易操作等优点,在农作物种子质量检测中被广泛使用。
荧光毛细管电泳技术将荧光检测的高灵敏度和毛细管电泳的高效性结合,利用电泳和电渗流的电动力学原理,在毛细管中灌满胶分离PCR产物,检测荧光信号,数据软件分析系统将其转化成峰图,显示试验结果。
该技术自动化程度高,检测通量高,试验操作简单,数据统计程序化,避免人为估算的主观性,结果精确到1bp[1],直接显示扩增产物片段的大小,实现了指纹图谱与碱基序列长短之间的对应转换,易于实验室间结果比对,常用GeneMapper 和GeneMarker分析SSR扩增片段[2]。
本文就试验中遇到的荧光信号强度、特异峰型的统计分析以及毛细管电泳仪运行和维护、试验结果与数据库比对等问题进行分析,为SSR分子标记荧光毛细管电泳检测农作物种子质量试验技术标准化提供参考。
拟,为所有专业方提供可控管、无破坏性、性价比高、低风险并允许反复运用的可行性方法。
BIM技术可以有效地提高施工技术水准,预防施工事故,减少施工成本浪费并缩短时程,强化施工过程中决策、控管能力。


一、无样品峰出现A、检查电流是否稳定:①没有电流。
可能原因——毛细管堵塞或断裂。
解决方法——用水冲洗毛细管,并观察是否有水流出,若无水流出请拆下卡盒检查毛细管两端和窗口是否断裂;毛细管没有断裂的话可以用水反向高压冲洗以试图解决此问题。
缓冲溶液需要过滤,将样品过滤或者离心去除其中的颗粒。
②电流波动很大,直至几乎消失。
可能原因——缓冲溶液中有气泡产生或者区带中样品析出。
解决方法——将缓冲溶液超声脱气,如果还有此现象发生,则可能是样品区带有析出,可以通过降低样品浓度/延长ramptime来试图解决这一问题;对于在缓冲溶液中溶解度不高的样品则需要在缓冲溶液中加入添加剂以解决此问题。
③电流初始值较小,后逐渐增大。
可能原因——样品进样量过大。
解决方法——减少进样量,通常进样参数设置在0.5psi,5sec 左右。
④电流正常。
可能原因:a样品浓度过低:使用高浓度样品测试,如果无法解决则有可能是以下其他原因。
b检测波长设置不正确:请确认被分析物的特征吸收,检查方法中的检测波长设置。
c分离极性错误:对于蛋白样品,请注意蛋白在分离条件下其PI及所带电荷;对于核酸样品,通常条件下会带负电荷。
d样品在毛细管内壁吸附:对于蛋白及核酸样品应尽量采用涂层毛细管分离,或采用极端pH条件或动态涂层防止样品吸附。
e光学检测器或光纤损坏:进行标准样品的测试,如果没有对应的结果出现,则有可能存在硬件问题,请联系工程师。
B、检查毛细管窗口,是否有透明窗口:可能原因——忘记开毛细管窗口或窗口位置不正。
解决方法——重新开毛细管检测窗口,或将窗口调整到正确位置。
二、样品峰出现拖尾可能原因——样品在毛细管内壁吸附。
解决方法——对于蛋白及核酸样品应尽量采用涂层毛细管分离,或采用极端pH条件或动态涂层防止样品吸附。
三、样品峰形不对称A、检查毛细管入口:可能原因——毛细管入口切口不平齐。
解决方法——重新切割毛细管入口,注意毛细管切割方法,不可以用力过猛或反复刮擦。
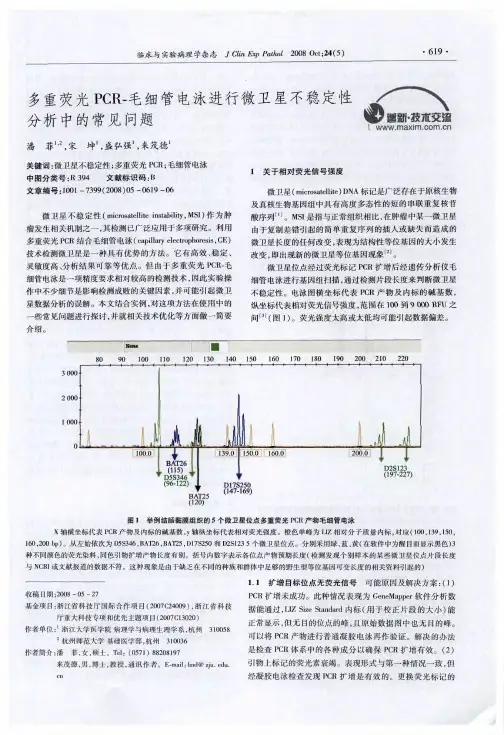

电泳中断原因分析毛细管电泳(capillary electrophoresis,CE)又称高效毛细管电泳(high perform ance capillary electrophoresis,HPCE),是一类以毛细管为分离通道、以高压直流电场为驱动力的新型液相分离技术。
毛细管电泳实际上包含电泳、色谱及其交叉内容,它使分析化学得以从微升水平进入纳升水平,并使单细胞分析,乃至单分子分析成为可能。
毛细管电泳技术的不断发展,毛细管电泳仪也越来越多的得到了大家的认同,但往往我们在试验的过程中经常会遇到一些问题,如样品的重现性,运行时的电流波动,阻碍电泳进一步发展的重要问题,及时的解决电泳的相关问题,对电泳的发展乃是分析化学的未来有这重要的现实意义。
毛细管电泳是在高压的作用下,以毛细管为分离通道,柱子中充满着缓冲溶液,毛细管两端浸泡在缓冲溶液的小瓶中,如图1所示,这样就构成了回流,对应的回流也就有了电流,但往往在试验的过成中电流并不像我们想的那样稳定,甚至有断流的现象,接下我们分析一下产生这种现象可能的原因。
图1 毛细管电泳仪示意图可能导致断流的原因:1、毛细管电泳柱子1.1柱子断裂毛细管柱子外壁涂有聚亚酰胺涂层,有很好的韧性,一段这层涂层脱落,毛细管柱子将失去原有的韧性,变得很脆,很容易折断。
取毛细管柱子时千万不要用利器划柱子,否则柱子很容易在划痕处断开。
毛细管柱子断裂后,缓冲溶液从断裂处流出色谱柱子,进而不能构成回路,从而导致电流迅速下降,致使电流中断,分离分析中断。
解决方法:更换柱子(平时更换柱子特别注意,不要用利器划到柱子)图2 断了柱子的毛细管电泳示意图1.2毛细管电泳柱子堵了常用的毛细管电泳柱子内径有50微米、75微米及100微米,内径很小,如果我们在试验的过场中不注意缓冲溶液及样品的纯度,或是使用了粘度很大的缓冲溶液,很容易堵死毛细管柱子,造成电流的中断。
图3柱子被堵后毛细管电泳示意图解决方法:压力反向冲洗柱子、样品及缓冲溶液使用前过滤2、气泡问题2.1缓冲溶液中的气泡我们在配置缓冲溶液时,所选用的水及在配置的过程中会使缓冲溶液溶解少量的气体,当这部分缓冲溶液进入到毛细管柱子内部时,两端加高压,运行一段时间会是缓冲溶液中的气体溢出,使柱子内产生气泡,从而使电流中断。



毛细管电泳实验报告(含预习报告)
系别____________ 学号 ________ 姓名 _________
组别 ____ 同组姓名 ___________________
实验日期年月日星期____ 得分____
1 实验目的
2 实验原理
3 预习报告
3.1请试回答以下问题
试列举影响电渗流(大小和/或方向)的几个主要因素:___________________;
决定离子电泳淌度的参数:,,。
3.2 查找下列物质的pKa值,并初步判断出峰顺序
苯甲醇:,苯甲酸:,水杨酸:,对氨基苯甲酸:
预测出峰顺序:
3.3进样前使用三种溶液对毛细管冲洗的目的分别是什么?
3.4 试列举毛细血管电泳的主要优势和劣势。
4 实验仪器及条件
仪器型号:
毛细管总长:毛细管有效长度:检测波长:
分析电压:进样压力:进样时间:
5 实验内容
6 数据处理
6.1 数据记录
6.2 各组分浓度计算(单点外标定量法)
计算公式:
6.3 各组分淌度计算
表观淌度计算公式:
有效淌度计算公式:
7 结论及讨论
(试讨论:1.判断在另外两种缓冲液下,各个峰的归属,并对各个组分迁移时间的变化做出合理分析和讨论;2. 判断哪个组分可以作为电渗流标记物,并试根据淌度结果说明之)
8 参考文献
(附:电泳谱图)。

综 述影响毛细管电泳分离生物大分子的因素刘 勇1,2 王 荣1,2 高 岚2 贾正平1,2 辛晓婷2 谢 华1 马 骏1(1.兰州军区兰州总医院临床药理基地,兰州,730050;2.兰州大学生命科学学院,兰州,730000) 摘 要 目前毛细管电泳在分离方面的应用越来越广泛,但重复性一直是制约其应用的主要问题。
随着毛细管电泳技术的发展,影响毛细管电泳结果的因素也发生着变化。
本文对影响毛细管电泳分离生物大分子的因素进行综述,希望对使用毛细管电泳的使用者者有所帮助。
关键词 毛细管电泳 分离 影响因素基金项目:国家自然科学基金项目(No .20775089)。
作者简介:刘勇,男,1984年6月出生,硕士,辽宁抚顺人,主要从事毛细泳和分子生物学研究工作。
E -mail :liuy .8406@ 毛细管电泳(CE )现在已经广泛应用于分子生物学、分子医学和生命科学等领域,近年来在生物大分子分离方面应用更为突出。
CE 顾名思义,就是在一根毛细管中完成电泳过程,这种方法的灵敏度是非常高的,但就是这种高灵敏性对于条件的改变特别敏感,影响了重复性。
如果对其条件进行严格控制并仔细操作的话,CE 确实是一种非常高效、快速、简易的检测方法。
本文查阅20年来毛细管电泳相关文献,对影响CE 结果的因素进行综述,以期对毛细管电泳的使用者有所帮助。
1 检测器检测器是毛细管电泳仪的关键部件之一,它的作用是将毛细管柱流出物中样品组分的含量随时间的变化转化成易测量的电信号。
用记录仪记录电信号,得到样品组分的电泳图。
根据组分峰的位置、大小和形状进行定性、定量分析,以及分离优劣的判断。
目前使用最广的两种检测器为紫外检测器和荧光检测器。
紫外吸收检测器是毛细管电泳最早选用的检测器,它因结构简单、操作方便、灵敏度适中而应用广泛。
但其最大缺点是检测光程短,难以进一步提高检测的灵敏度,最低检测限一般在10-5~10-6m ol /L 之间。
荧光检测器特别是激光诱导的荧光检测器(LIFD ),是在普通荧光检测器的基础上发展起来的,从1987年开始才有应用于毛细管电泳的报道[1],因其灵敏度极高、性能可靠、易操作、使用成本低而迅速得到广泛的应用[2],尤其适合应用于氨基酸、DNA 片段等生化样品的检测。

高效毛细管电泳仪实验过程中常见问题解答序号现象可能原因纠正措施1电源开启后毫无反应1.电源没接通2.保险丝已断将仪器插头从电源插座中取出,确认插座里有电,检查保险丝,更换电源线。
2 电流指示为零电极断掉,弯曲或没有插对更换、校正、重插。
3仪器实验过程中电流不稳或电流指示远底于正常值,但电压值正常1.毛细管堵塞2.毛细管中无缓冲溶液3.毛细管破裂4.电压未加5.仪器参数设定错误6.柱内有大气泡7.用水做溶剂配样,在压力进样时将一部分水压入缓冲溶液中重新用HCl、NaOH清洗毛细管或剪去进样端小段毛细管,如果仍旧不能清洗,更换毛细管。
检查毛细管是否浸在缓冲溶液中。
更换毛细管。
检查电压值。
重新设定仪器参数。
重新注入缓冲溶液。
用稀释的运行缓冲溶液代替水配样。
4 电流指示过高毛细管柱过热用干净缓冲溶液冲洗毛细管柱,使之冷却。
5电流指示不断变化1.毛细管内有许多小气泡2.环境温度过高或过底,通风不好重新注入缓冲液。
调整环境温度,最好使用控温装置。
6恒压操作时电压显示值闪动电流设置过底重新设置电流值。
7电泳谱图上没有出现峰1.样品浓度太低或体积太小2.样品被管壁严重吸附3.毛细管检测窗与检测器光路未对准4.信号线连接错误增加样品浓度或样品体积。
对毛细管壁进行修饰处理。
取出毛细管,重新安装。
检查信号线连接情况。
8基线漂移或噪声较大1.毛细管被污染或未清洗2.毛细管柱的光学窗口被污染3.有小气泡4.操作电压太高5.缓冲溶液浓度太大6.缓冲溶液或样品中有小颗粒物质7.毛细管有很小裂缝8.检测器灯源老化9.接地问题10.检测器参数设定错误11.数据系统设定错误用0.5mol/LNaOH溶液、水、缓冲液清洗毛细管。
取下检测池,清洗光学透镜。
缓冲溶液脱气。
降低操作电压。
降低冲溶液浓度。
过滤缓冲溶液或样品溶液并检查缓冲溶液与样品是否发生作用。
更换毛细管。
更换灯源。
将检测仪和电泳仪连接在同一电源上。
检查检测器参数设定值。
重新设定数据处理参数。

毛细管电泳技术的优化及应用研究毛细管电泳技术是一种基于毛细管内分离某些电荷、形状或大小不同的分子物质的方法,这项技术已经广泛应用于治疗癌症、检测药物以及环境监测等方面。
随着科技的不断发展,毛细管电泳技术的优化及其应用研究也在不断深入进行。
一、毛细管电泳技术的优化尽管毛细管电泳技术在分析某些更复杂的分子物质方面取得了成功,但其检测灵敏度和分离效率还有需要改进的地方。
在优化毛细管电泳技术时,对于改善检测灵敏度和分离效率的方法,我们需要考虑如下几个方面:1、分离柱质量的选择毛细管电泳是一种基于柱填料的方法。
因此,分离柱的质量对于毛细管电泳分析的影响很大。
2、检测器的选择检测器的敏感度对于毛细管电泳分析结果的准确性和稳定性有着极大的影响。
在选择检测器时,需要考虑其灵敏度、响应时间和稳定性等因素。
3、毛细管的制备和修整毛细管的制备和修整不仅会影响毛细管电泳分析过程中的分离效率和检测灵敏度,还会影响分离柱的寿命。
4、电泳缓冲液的选择电泳缓冲液对于毛细管电泳的分离效率、灵敏度和稳定性等方面都有很大的影响。
因此,在选择电泳缓冲液时需要考虑其PH值和离子强度等因素。
二、毛细管电泳技术在药物检测中的应用毛细管电泳技术在药物分析和检测方面得到了广泛的应用。
药物分析中,毛细管电泳技术可以用于对药物的定量和质量分析。
例如,对于一些具有毒副作用,易于滥用或者难以检测的药物,如海洛因和可卡因等,毛细管电泳技术可以快速,准确地检测出药物的存在,并定量分析其浓度。
此外,毛细管电泳技术在药物分析和检测方面还可以用于药物无害性研究、新药研发以及药物代谢研究等方面。
三、毛细管电泳技术在环境监测中的应用毛细管电泳技术在环境监测领域的应用也越来越广泛。
它可以用于快速、准确地检测环境中某些污染物的存在。
毛细管电泳技术可以用于检测水、土壤和大气中的环境污染物,如重金属、农药、环境荷尔蒙等。
除了可以检测这些污染物存在的问题外,毛细管电泳技术还可以帮助我们研究产生这些污染物的原因。
毛细管分析常见问题的解决一、峰丢失可能的原因及应采用的排除方法1.注射器有毛病,用新注射器验证。
2.未接入检测器,或检测器不起作用,检查设定值3.进样温度太低,检查温度,并根据需要调整4.柱箱温度太低,检查温度,并根据需要调整5.无载气流,检查压力调节器,并检查泄漏,验证柱进品流速6.柱断裂,如果柱断裂是在柱进口端或检测器末端,是可以补救的,切去柱断裂部分,重新安装二、前沿峰1.柱超载,减少进样量2.两个化合物共洗脱,提高灵敏度和减少进样量,使温度降低10~20度,以使峰分开3.样品冷凝,检查进样口和柱温,如有必要可升温4.样品分解,采用失活化进样器衬管或调低进样器温度三、拖尾峰1.进样器衬套或柱吸附活性样品:更换衬套。
如不能解决问题,就将柱进气端去掉1~2圈,再重新安装2.柱或进样器温度太低:升温(不要超过柱最高温度)。
进样器温度应比样品最高沸点高25度3.两个化合物共洗脱:提高灵敏度,减少进样量,使温度降低10~20度,以使峰分开4.柱损坏:更换柱5.柱污染:从柱进口端去掉1~2圈,再重新安装四、只有溶剂峰1.注射器有毛病:用新注射器验证。
2.不正确的载气流速(太低):检查流速,如有必要,调整之3.样品太稀:注入已知样品以得出良好结果。
如果结果很好,就提高灵敏度或加大注入量。
4.柱箱温度过高:检查温度,并根据需要调整5.柱不能从溶剂峰中解析出组分:将柱更换成较厚涂层或不同极性6.载气泄漏:检查泄漏处(用肥皂水)7.样品被柱或进样器衬套吸附:更换衬套。
如不能解决问题,就从柱进口端去掉1~2圈,并重新安装五、宽溶剂峰1.由于柱安装不当,在进样口产生死体积:重新安装柱。
2.进样技术差(进样太慢):采用快速平稳进样技术。
3.进样器温度太低:提高进样器温度。
4.样品溶剂与检测相互影响(二氯甲烷/ECD):更换样品溶剂。
5.柱内残留样品溶剂:更换样品溶剂6.隔垫清洗不当:调整或清洗7.分流比不正确(分流排气流速不足):调整流速六、假峰1.柱吸附样品,随后解吸:更换衬管,如不能解决问题,就从柱进样口端去掉1~2圈,再重新安装。
无样品峰出现
A、检查电流是否稳定:
①没有电流。
可能原因——毛细管堵塞或断裂。
解决方法——用水冲洗毛细管,并观察是否有水流出,若无水流出请拆下卡盒检查毛细管两端和窗口是否断裂;毛细管没有断裂的话可以用水反向高压冲洗以试图解决此问题。
缓冲溶液需要过滤,将样品过滤或者离心去除其中的颗粒。
②电流波动很大,直至几乎消失。
可能原因——缓冲溶液中有气泡产生或者区带中样品析出。
解决方法——将缓冲溶液超声脱气,如果还有此现象发生,则可能是样品区带有析出,可以通过降低样品浓度/ 延长ramp time 来试图解决这一问题;对于在缓冲溶液中溶解度不高的样品则需要在缓冲溶液中加入添加剂以解决此问题。
③电流初始值较小,后逐渐增大。
可能原因——样品进样量过大。
解决方法——减少进样量,通常进样参数设置在0.5psi,5sec 左右。
④电流正常。
可能原因:a 样品浓度过低:使用高浓度样品测试,如果无法解决则有可能是以下其他原因。
b 检测波长设置不正确:请确认被分析物的特征吸收,检查方法中的检测波长设置。
c 分离
极性错误:对于蛋白样品,请注意蛋白在分离条件下其PI及所带
电荷;对于核酸样品,通常条件下会带负电荷。
d样品在
毛细管内壁吸附:对于蛋白及核酸样品应尽量采用涂层毛细管分
离,或采用极端pH条件或动态涂层防止样品吸附。
e光学检测器或光纤损坏:进行标准样品的测试,如果没有对应的结果出现,则有可能存在硬件问题,请联系工程师。
B、检查毛细管窗口,是否有透明窗口:
可能原因一一忘记开毛细管窗口或窗口位置不正。
解决方法一一重新开毛细管检测窗口,或将窗口调整到正确位置。
二、样品峰出现拖尾
可能原因一一样品在毛细管内壁吸附。
解决方法一一对于蛋白及核酸样品应尽量采用涂层毛细管分离,或
采用极端pH条件或动态涂层防止样品吸附。
三、样品峰形不对称
A、检查毛细管入口:
可能原因一一毛细管入口切口不平齐。
解决方法一一重新切割毛细管入口,注意毛细管切割方法,不可以
用力过猛或反复刮擦。
B、检查更改样品溶剂:
可能原因——缓冲溶液与样品溶液电解率差别过大。
解决方法一一可采用缓冲溶液作为样品溶剂
四、样品峰过宽
降低样品进样量:
a有改善:可能原因一一样品浓度太大。
解决方法一一可降低样品浓度或减少进样量。
b无改善:可能原因一一样品本身性质不均一。
解决方法一一此原
因主要是针对蛋白样品,小分子样品发生的概率较低。
五、迁移时间不稳定
①出峰时间不稳定且无规律:可能原因一一缓冲溶液与毛细管内壁
平衡较慢。
解决方法一一每次样品运行之间避免用NaOH中洗毛细管。
②出峰时间依次后延:可能原因一一样品中有物质易发生吸附。
解决方法——首先在每次样品运行之前用0.1N NaOH短时间冲洗毛细管,看迁移时间能否重复,如果效果不佳请使用涂层毛细管来改善结果或者将样品再次处理,以去除样品中易吸附的组分。
六、峰形和出峰时间不稳定
更换缓冲溶液种类:
①峰形和时间稳定:可能原因一一缓冲溶液不稳定,易电解,或者是样品在原缓冲溶液条件下不稳定。
解决方法一一更换其它种类的缓冲溶液。
②峰形和出峰情况仍不稳定:可能原因一一样品中物质易在毛细管内壁吸附。
解决方法一一可采用涂层毛细管来克服此问题,或对样品进行前处理。
七、无法加气压
检查瓶塞是否盖紧或更换瓶塞。
①更换后问题解决。
可能原因a瓶塞老化,失去弹性。
解决方法一
—请购买新的瓶塞,并注意瓶塞不可烘干,只能自然晾干。
b瓶颈处有液体,导致瓶塞易滑。
解决方法——向瓶中装液体时不要过满,也不要沾湿瓶颈。
②若瓶塞无问题,可先采用真空方式,若真空方式可以使用,但压力仍不可用。
可能原因一一压力系统问题。
解决方法一一请联系工程师。
八、迁移时间可重复但峰面积重复性不好
重新切割毛细管两端。
①若问题解决。
可能原因一一毛细管入口处不平。
解决方法一一重新切割毛细管入口,注意毛细管切割方法,不可以用力过猛或反复
刮擦②问题依然存在,尝试电动进样。
可能原因——若电动进样可保持重现,则压力控制或气路存在问题。
解决方法——请联系工程师。
九、毛细管或电极易折断
①检查瓶盖是否老化。
可能原因——瓶盖老化。
解决方法——购买新的瓶盖。
②电极是否不垂直。
可能原因电极不垂直。
解决方法将电
极拉直。
十、谱图基线不稳定
①先用0.1N NaOH中洗毛细管,然后不进样运行原方法。
a 基线改善。
可能原因——则为样品中物质有吸附。
解决办法——重新预处理样品或更改电泳条件。
b基线未改善。
可能原因一一毛细管内有强烈吸附,或缓冲体系不稳定。
解决办法——更换新的毛细管,不进样,只运行缓中,观察基线情况。
②更换新毛细管后基线仍然不稳,可采用Run Buffer A测试。
a基线改善。
可能原因一一缓冲溶液与毛细管内壁平衡较慢。
解决办法——更换缓中溶液种类,若基线变化对检测无干扰,可保持原来电泳条件。
b 基线未改善。
可能原因——检测光源或其他光学器件不正常。
解
决办法请联系工程师。