上机-第二代测序中的数据分析-转录组
- 格式:pdf
- 大小:2.19 MB
- 文档页数:40

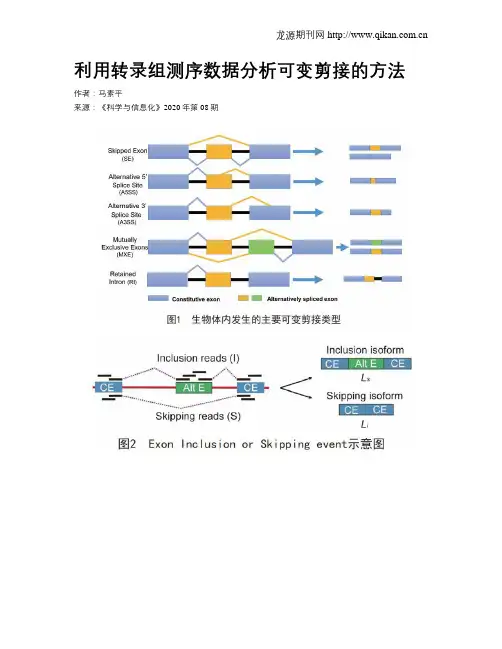
利用转录组测序数据分析可变剪接的方法作者:***来源:《科学与信息化》2020年第08期摘要可变剪接是调节基因表达和产生蛋白组多样性的重要因素,同时参与调控细胞分裂、分化及凋亡等重要生物学过程,异常的可变剪接多与人类疾病有关。
随着新一代测序技术和生物信息学的快速发展,以及先进计算方法的提出,使得我们对可变剪接有了深入的认识。
并且基于剪接机制对于病的靶向药物设计,已得到了有效的临床治疗效果。
本文主要阐述了近年来基于二代测序技术开发的几种识别可变剪接的计算方法,并对未来的发展方向进行展望。
关键词可变剪接;二代测序技术;生物信息学;分析工具可变剪接,又称选择性剪接(Alternative Splicing,AS),是真核生物基因表达的普遍调节机制,是指一个前体mRNA经过不同的剪接形式产生多种不同剪接异构体的过程。
在1978年,Walter Gilbert提出了内含子和外显子命名[1],不同外显子组合产生特异的异构体。
二代测序技术的迅速发展极大地推动了人类对可变剪接的认识。
现有数据表明,人类大约有92%-94%的基因都会经历某种程度的可变剪接行为,并且在20000多种人类蛋白编码基因中,约37%的基因会编码产生不同的蛋白亚型,这表明可变剪接增加了蛋白质组的多样性和复杂性[2]。
AS对基因的功能起着重要调控作用,同一基因的不同亚型可能参与不同的生物学过程。
例如p53抑癌基因(TP53)在DNA受损细胞的调控中起着核心作用,然而其Δ133β亚型则可以抑制全长p53β亚型5和6从而诱导肿瘤细胞的凋亡[3]。
另外AS几乎参与了所有生物学过程,包括调节细胞的分裂和凋亡、神经系统的发育以及细胞对抗多种环境因素做出的免疫应激反应等[4]。
另一方面,AS的异常调节还与多种遗传性疾病和恶性肿瘤相关,包括神经退行性疾病、心血管疾病和代谢状况等。
据报道,与SNP相关的遗传性疾病多达一半是由于剪接受损引起的[5]。
AS的异常调节对癌症的发生发展有重要的作用,为疾病的发展提供了可能的新颖治疗靶标和生物标志物的来源,而AS位点的预测可以为药物设计提供很好的分子基础。

《第二代测序技术的发展及应用》篇一一、引言随着人类对生命科学研究的不断深入,测序技术作为生命科学研究的重要手段之一,其发展历程也经历了多次重大突破。
其中,第二代测序技术作为当前应用最广泛的测序技术之一,其发展及应用对于生命科学研究、医学诊断、药物研发等领域产生了深远的影响。
本文将重点介绍第二代测序技术的发展历程、原理、应用及未来展望。
二、第二代测序技术的发展历程及原理1. 发展历程第二代测序技术,又称高通量测序技术,自2005年问世以来,经历了从初期的小规模应用到现在的大规模商业化应用的历程。
其发展主要得益于大规模并行测序技术的突破和生物信息学技术的进步。
2. 原理第二代测序技术基于大规模并行测序原理,通过将待测序列的DNA分子进行大规模的扩增和测序,从而实现高通量、高精度的测序。
其主要步骤包括DNA文库构建、桥式PCR扩增和碱基识别等。
三、第二代测序技术的应用1. 生命科学研究第二代测序技术在生命科学研究中得到了广泛应用。
例如,通过对基因组、转录组等数据的测序和分析,研究人员可以了解基因的表达、变异、互作等信息,为基因疾病的研究提供重要依据。
此外,第二代测序技术还可以用于物种进化分析、基因组拼接等领域。
2. 医学诊断第二代测序技术在医学诊断中也有着重要的应用。
例如,通过对患者肿瘤组织的基因组测序,可以了解肿瘤的基因突变情况,为肿瘤的个性化治疗提供重要依据。
此外,第二代测序技术还可以用于病原体检测、遗传病诊断等领域。
3. 药物研发第二代测序技术在药物研发中也具有重要作用。
通过对药物的靶点进行基因组或转录组分析,可以了解靶点的结构和功能信息,为新药设计和研发提供重要参考。
此外,第二代测序技术还可以用于药物临床试验中患者入组标准的制定等环节。
四、第二代测序技术的挑战与展望1. 挑战尽管第二代测序技术已经取得了巨大的成功,但仍面临着一些挑战。
例如,随着测序数据的不断增长,如何进行高效的数据分析和解读成为了一个重要问题。


二代测序分析流程Next-generation sequencing (NGS) has revolutionized the field of genomics by allowing researchers to rapidly sequence large amounts of DNA and RNA. 二代测序(NGS)已经彻底改变了基因组学领域,使研究人员能够快速测序大量的DNA和RNA。
This technology has enabled the analysis of entire genomes, transcriptomes, and epigenomes, providing a wealth of data that can be used to study genetics, disease, and evolution. 这项技术使得对整个基因组、转录组和表观基因组的分析成为可能,为研究遗传学、疾病和进化提供了大量的数据。
One of the key challenges in NGS is the analysis of the data generated, which requires a complex and multi-step process to extract useful information. 二代测序面临的关键挑战之一是分析生成的数据,这需要复杂且多步骤的过程来提取有用的信息。
The NGS analysis pipeline typically involves several key steps, including quality control, read mapping, variant calling, and downstream analysis. 二代测序分析流程通常包括几个关键步骤,包括质量控制、读片段比对、变异检测和下游分析。

第1篇一、背景随着高通量测序技术的发展,转录组分析已成为研究基因表达调控和基因功能的重要手段。
本报告针对某研究项目中的转录组数据进行分析,旨在探究该物种在不同生长阶段的基因表达差异及其生物学意义。
二、实验方法1. 样本采集:在研究项目中对不同生长阶段的样本进行采集,包括幼年期、成熟期和衰老期。
2. RNA提取:采用TRIzol法提取样本总RNA,并进行质量检测。
3. cDNA文库构建:采用SMART-seq2技术构建cDNA文库。
4. 转录组测序:使用Illumina HiSeq平台进行转录组测序。
5. 数据分析:采用HTSeq-count软件对测序数据进行定量,利用DESeq2进行差异表达分析,并使用GSEA进行基因集富集分析。
三、结果与分析1. 数据质量评估:测序数据经过质量控制后,得到有效数据量约为100亿个reads。
2. 基因表达差异分析:在幼年期、成熟期和衰老期三个阶段,共检测到差异表达基因(DEGs)1000个,其中上调基因600个,下调基因400个。
3. 基因功能富集分析:通过对差异表达基因进行GO和KEGG富集分析,发现与细胞周期、代谢、信号转导等生物学过程相关的基因富集显著。
4. 蛋白质互作网络分析:构建DEGs的蛋白质互作网络,发现某些关键基因在转录调控和信号转导过程中发挥重要作用。
四、结论本研究通过对某物种不同生长阶段的转录组数据进行分析,揭示了该物种在不同生长阶段的基因表达差异及其生物学意义。
研究结果为进一步研究该物种的生长发育机制提供了重要参考。
五、展望1. 深入挖掘差异表达基因的功能:通过实验验证差异表达基因的功能,揭示其在生长发育过程中的作用。
2. 分析差异表达基因的调控网络:进一步研究差异表达基因的调控网络,揭示基因表达调控的分子机制。
3. 结合表观遗传学分析:探究表观遗传学因素对基因表达的影响,为研究基因表达调控提供新的思路。
4. 探索转录组分析在植物育种中的应用:将转录组分析应用于植物育种,提高育种效率。

2019,38(1)河南大学学报(医学版)•67・文章编号:1672-7606(2019)01-0067-10基于二代测序技术的转录组测序生物信息分析汤冬-张国森2,赵晓芳回1.江苏苏博生物医学股份右限公司,江苏宿迁22380();2.河南犬学基础恢学院医学生物信息学研究所细胞信号转导实验室,河南开封475004摘要:随着二代测序技术和生物岱息学的发展,越来越多的科研人员通过转录组测序(RNAseq)研究基因表达调控、疾病发生机制和遗传育种上的问题面对测序所产生的大量数据.生物信息学分析策略对于数据的解读显得尤为車:要本文结合不同RNA(mRNA J.ncRNA、miRNA和eireRNA)的特点,对转录组测序中的儿类分析流程及其所涉及的软件和数据库分别做简要的介绍.为RNAseq的生信分析研究提供参考关键词:二代测序;转录组;测序;个物信息中图分类号:Q786文献标志码:ABioinformatics analysis of transcriptome sequencing based on next generation sequencingTANG Dong1,ZHAGN Guo sen2,ZHAO Xiaofang"31.Jiangsu Superbio Life Science Co.Ltd..Suqian223800.China:2.Institute of Biomt'dical Informatics.Cell Sigiuil Transduction 1l aboratory,School of Basic Medical Sciences of Henan University,Kaifeng475004.ChinaAbstract:With the rapid(lev(4opinent of the nexl gen e ration sequencing(N(;S)and bioinfonnatics recently,more and more resriirclirrs devote to tlx*tianscriplome studies through RNAseq.RNAseq can be used in the study of gene expression and regulation.the mechanism of disease,genetic breeding.Facr to a large amount of sequencing data,the amdytic strategy^of the (laki by bioinfonntitits is piirticularly important.In this review,we described several RNAseq analysis processes according to the features of different types of RNA(mRNA.LncKX A.rniRN A,circRNA),and also discussed th<*software and databases used in the processes briefly,which would provide a reference to the analysis of RNAseq.Key words:next gen(*ratioii sequencing;transcriptome;sequencing;bioinfonnatics第一代测序技术是以1970年代的Sanger法为代表的末端终止法测序技术「,广泛应用于人类基因组计划:随着测序技术的飞速发展.到2005年时,新一代的测序技术已被研发出来',通常被称为二代测序技术或者下一代测序技术(Next Generation Sequencing,NGS)由于解决了Sanger测序法通M低的缺点,二代测序技术采用边合成边测序的思想,可以一次性对上百万条核酸分子进行测序,得到几百亿个碱基序列,所以也被称为高通量测序(High-Throughput Sequencing,HTS)由于其高通量的特点,一次测序所获得的数据量可以覆盖到整个基因组或者转录组,使得研究人员通过测序对某个物种的基因组或者转录组做细致全貌的分析变得更加快捷和容易因此,越来越多的科研人员使用二代测序技术来分析生物学问题,致使生物学研究新领域不断被发掘目前.二代测序广泛应用在生收稿日期:2018-02-17基金项目:国家自然科学基金青年项目(31800431)作者简介:汤冬(1991-),男,作物遗传育种硕士,研究方向:分子遗传学回通信作者:赵晓芳(1982-),女,硕士.工程师,研究方向:生物信息学,E-mail:zhaoxiaofang@•&8•Journal of Henan Lniversitv(Me(li(al Science)2019,38(1)物的全基因组测序、转录组测序'、染色体免疫共沉淀测序*、I)NA甲基化测序5等方面可以说随着二代测序技术的诞生,人们对于基因组学的研究进入了一个全新的时代——后基因组学时代转录组学是继基因组学之后,快速发展t L应用最为广泛的研究技术遗传中心法则°认为,遗传信息从DNA传递给RNA.再从RNA传递给蛋白质.从而完成遗传信息的转录和翻译,因此,人类基因组i I"划完成后,研究方向也从基内测序转移到基因功能的注释上转录组作为连接承载遗传岱息的基因组和生物功能的蛋门组间的纽带,存在着许多木知的领域.有着许多值得研究的地方转录组在广义匕的定义是指某一生理条件卜-,细胞内所有转录产物的集合,包含mRNA、rRNA、tRNA和noncoding RNA 等;狭义上是指所有参与了翻译蛋门质的mRNA的集合根据RNA是否具冇编码蛋门质的能力,RNA 可以分成两大类:编码RN A(coding RNA)和非编码RNA(noncoding RNA)编码RNA即niKNiA(messenger RNA),属于狭义转录组RNA,是目前研究最多的一类RNA mRNA能够编码蛋白质,是遗传信息到生物功能的直接体现非编码RNA长期以来由于其不具备编码蛋口质的能力而被人们忽视. ENCODE计划研究表明,人类基因组大约75%的序列能转录成RNA,其中74%的是非编码RNA序列"。

转录组测序概述及实验分析流程(分享)⼀、转录组测序概述转录组是特定物种、组织或细胞类型转录的所有RNA(转录本)的集合,包括mRNA和⾮编码RNA(Non-coding RNA,⾮编码RNA⼜包括:tRNA,rRNA,snoRNA,microRNA,piRNA,lncRNA等。
通过⽐较转录组或基因表达谱的研究以揭⽰⽣物学现象或疾病发⽣的分⼦机制是⾼通量组学研究的⼀个常⽤策略。
利⽤⾼通量测序技术研究转录组在全⾯快速得到基因表达谱变化的同时,还可以通过测定的序列信息精确地分析转录本的cSNP(编码序列单核苷酸多态性)、可变剪接等序列及结构变异,另外对于检测低丰度转录本和发现新转录本具有其独特的优势。
⼆、研究转录组⽅法有哪些⽬前研究转录组的⽅法主要三种:1. 基于杂交技术的cDNA芯⽚和寡聚核苷酸芯⽚2. 基于sanger测序法的SAGE (serial analysis of gene expression)、LongSAGE和MPSS(massively parallelsignature sequencing)3. 基于第⼆代测序技术的转录组测序,⼜称为RNA-Seq。
三、转录组测序有什么样的样品要求?(1)样品纯度要求: OD值应在1.8⾄2.2之间;电泳检测28S:18S⾄少⼤于1.8。
(2)样品浓度: totalRNA浓度不低于400ng/µg。
(3)total RNA样品请置于-20℃保存;请提供totalRNA样品具体浓度、体积、制备时间、溶剂名称及物种来源。
请同时附上QC数据,包括电泳胶图、分光光度或Nanodrop仪器检测数据。
(4)样品请置于1.5 ml管中,管上注明样品名称、浓度以及制备时间,管⼝使⽤Parafilm封⼝。
建议使⽤⼲冰运输,并且尽量选⽤较快的邮递⽅式,以降低运输过程中样品降解的可能性。
四、转录组测序需要多⼤的测序量才能得到有意义的结果?转录组测序前,需要对物种转录组的⼤⼩进⾏评估,评估⽅法如下:(1)对于有reference genome的物种,可以分析基因组信息,统计编码基因的个数,及其碱基数,从⽽估计物种转录组的⼤⼩,另外可以查询相关或相近物种转录组研究的⽂献,作为参考。

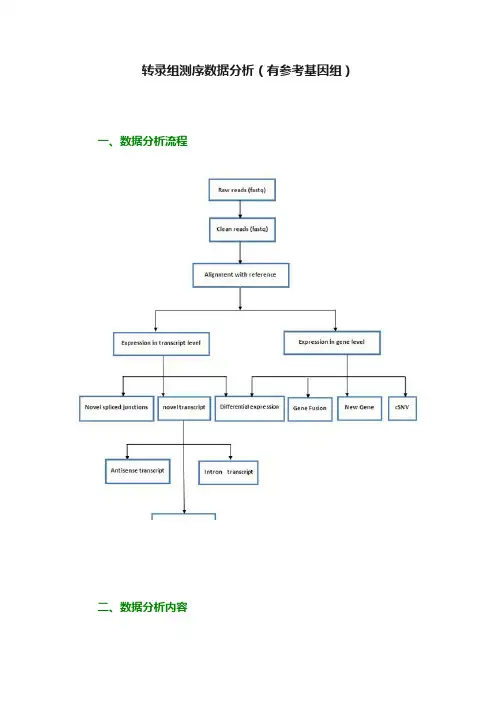
转录组测序数据分析(有参考基因组)一、数据分析流程二、数据分析内容1. 数据预处理目的:对原始测序数据进行一定程度的过滤。
原理:根据测序接头以及测序质量对原始的测序数据进行预处理,其中,测序质量Q与测序错误E之间的关系如下:结果:对预处理后质量以及碱基分布统计进行统计2. 比对基因组目的:将经过预处理的测序数据与参考基因组进行相似性比对。
原理:Burrower-Wheeler转换算法与splicing比对算法。
1)Burrower-Wheeler转换算法:由于测序数据量非常大,与整条基因组比对所需资源与时间是较为巨大的。
目前,我们采用Burrower-Wheeler(BWT)算法对基因进行建立索引、碱基压缩等过程,这样可以很大程度上加快比对速度,减少比对过程中所需资源。
2)splicing比对算法:即分段比对算法,当某条测序序列位于转录本剪切位点时,也就是这条序列同时属于两个外显子,如果将它与参考基因组进行比对,由于基因组两个外显子之间含有intron区,那么它将无法找到它合适的位置;但是应用分段比对算法就可以将这条测序序列分割变成多段子序列,然后应用这些段子序列与基因组进行比对,这样就可以找到它们真正的位置。
Vps28基因的一个分段比对的结果,蓝线连接的两端即为被分割的子序列,可见此种算法非常的适用于转录组测序。
结果展示:应用比对结果进行一些相关mapping统计,测序饱和度及测序5’,3’ bias统计。
Multi mapping,Unique mapping及Unique gene-body mapping统计。
饱和度分析,当reads达到一定测序量后,基因覆盖率基本达到饱和。
测序3’,5’偏好性统计,测序主要集中于基因bady区,两端偏向性较轻。
3. 基因表达水平研究目的:应用基因组比对结果进行基因定量。
原理:从指定物种基因模型(基因结构)中得到gene、exon、intron以及UTR等位置信息,通过基因组比对结果计算出在不用区域富集片段数目,然后应用RPKM/FPKM标准化公式对富集片段的数量进行归一化。

第二代测序的原理及其应用1. 前言随着DNA测序技术的发展,第二代测序技术的出现为科研人员和生物医药领域带来了革命性的变化。
本文将介绍第二代测序的原理及其在科研和生物医药领域的应用。
2. 第二代测序的原理第二代测序是相对于第一代测序而言的,其主要特点是高通量和快速测序。
相比第一代测序,第二代测序技术可以在短时间内完成大规模的DNA测序。
第二代测序的原理基本上是通过将DNA样本分子化,并通过扩增、固定和测序的过程来获得测序结果。
具体步骤如下:•DNA片段的制备:首先,DNA样本需要进行切割,生成适当长度的DNA片段。
•适配体连接:将DNA片段连接到适配体上,适配体上含有特定序列,用于扩增和固定DNA片段。
•DNA扩增:通过PCR反应,对连接好的DNA片段进行扩增,以增加测序的灵敏度。
•DNA固定:将扩增的DNA片段固定在测序芯片或流式细胞中,以便进行后续的测序反应。
•测序反应:通过各种不同的测序技术(如Illumina、Ion Torrent 等),对DNA片段进行测序,得到碱基序列。
•数据分析:通过计算机算法,将得到的碱基序列进行比对和分析,得到最终的测序结果。
3. 第二代测序的应用第二代测序技术的高通量和快速特性使其在科研和生物医药领域有着广泛的应用。
以下是第二代测序技术的一些主要应用:3.1 基因组学研究•通过对整个基因组的测序,可以帮助科研人员了解基因组的结构、功能和变异情况。
•基因组测序还可以用于研究不同物种之间的遗传差异,揭示物种的进化历史。
3.2 转录组学研究•转录组测序可以帮助科研人员了解特定组织或细胞中的转录活动。
•通过比较不同条件下的转录组数据,可以探索基因表达的调控机制。
3.3 蛋白质组学研究•第二代测序技术结合质谱分析,可以用于高通量的蛋白质组学研究。
•可以通过测序和质谱分析,研究蛋白质的翻译后修饰和亚细胞定位。
3.4 癌症基因组学研究•通过对肿瘤患者的基因组测序,可以寻找与癌症相关的突变。
实用总结:转录组测序和分析,你需要知道的都在这儿了转录组测序及分析技术可以解决新基因的深度发掘、低丰度转录本的发现、转录图谱绘制、可变剪接的调控、代谢途径确定、基因家族鉴定及进化分析等各方面的问题;成为了广大科研工作者备受青睐的高通量测序技术之一。
转录组研究的应用领域十分广泛,适合研究组织特异性的、不同生长发育的、逆境胁迫下的、侵染转基因的、性状突变等材料。
转录组是在某一特定发育时期或某一生理条件下,细胞或组织内所有转录产物的集合,包括mRNA、lncRNA、small RNA、circle RNA等。
因此做转录组测序理论上可以研究各种长度范围的RNA序列,目前的常规技术包括mRNA测序、lncRNA测序、smallRNA测序。
那么问题来了,研究转录组如何下手?1根据研究对象,选择相应的建库策略(1)mRNA:可以通过富集polyA的方式来调取mRNA,进行建库测序;(2)lncRNA或lncRNA+mRNA:可以通过去rRNA试剂盒去除rRNA后进行建库测序;(3)circle RNA:可以通过消化线性RNA,再去除rRNA后进行建库测序;(4)small RNA:采用sRNA的建库策略,对18-40nt范围的sRNA进行切胶富集后建库测序。
2根据研究目的,选择不同的测序策略(1)了解不同样品间基因或sRNA的表达差异:选择SE(single end)测序即可,测序量10M reads以上;(2)进行基因的可变剪切、挖掘新基因、对现有基因的注释进行优化、检测基因融合等结构方面的分析:选择PE(pair end)测序,测序量则根据物种基因集合的大小来决定。
3基于转录组测序的主流研究手段(1)RNA-seq denovo:基于序列组装,用于从头构建某物种的转录本序列;(2)RNA-seq resequencing:对于已有参考基因的物种,进行基因定量、基因可变剪切、基因融合、新基因检测等分析;(3)lncRNA-sequencing:主要研究lncRNA的表达量,预测新的lncRNA及其功能;(4)sRNA sequencing:主要研究和分析small RNA序列,特别是miRNA的表达情况,并预测novel miRNA,miRNA靶基因分析等。
转录组高通量测序2010-11-22 09:48(第二代高通量测序技术-454)转录组即特定细胞在某一功能状态下所能转录出来的所有RNA的总和,是研究细胞表型和功能的一个重要手段。
与基因组不同的是,转录组的定义中包含了时间和空间的限定。
同一细胞在不同的生长时期及生长环境下,其基因表达情况是不完全相同的。
罗氏GS-FLX-Titanium第二代高通量测序仪平均读长超过400bp,在测序读长上遥遥领先于其它第二代高通量测序仪,使其成为转录组学研究的首选测序平台,已被广泛应用于基础研究、临床诊断和药物研发等领域。
一、罗氏454测序技术在环境微生物生态多样性研究中的突出优势体现在:(1)测序序列长,便于聚类拼接,可以对转录本进行从头组装(de novo assembly)。
(2)测序通量高,可以检测到低丰度转录本信息。
(3)可以对无基因组参考序列的新物种进行转录组测序,发现新的转录本和亚型。
(4)实验操作简单、结果稳定,可重复性强。
无需进行克隆的文库构建,双链cDNA连接454接头后可以直接进行测序,实验周期短。
(5)测序数据便于进行生物信息分析,可以进行基因差异表达分析、鉴定基因的可变剪切以及预测新基因。
二、美吉公司在环境微生物生态多样性研究中的突出优势体现在:(1)拥有自主实验室和高通量测序平台,可以根据客户要求灵活安排实验,实验周期短,取样方便,质量可靠。
(2)技术人员经验丰富,可以稳定地进行总RNA的提取和双链cDNA的合成,可以根据顾客要求第一时间提供实验方案。
(3)有专业的生物信息团队和大型计算机,可以为客户提供个性化的生物信息分析服务。
(4)开放式实验室,参与式服务。
客户不但可以参与整个实验过程,而且可以参与生物信息分析,提供最为增值的售后服务。
三、服务流程(1)客户提供样本背景信息、实验目的和实验预期。
(2)美吉公司设计实验方案,提供测序深度建议和生物信息分析建议。
(3)客户认可实验方案,双方签订项目合作协议。