BLAST 和 FASTA 的应用
- 格式:pdf
- 大小:138.56 KB
- 文档页数:15

blast参数1. 什么是blast参数?BLAST(Basic Local Alignment Search Tool)是一种常用的生物信息学工具,用于比对两个或多个生物序列,以寻找相似性和同源性关系。
在进行BLAST比对时,我们可以设置一些参数来调整比对的敏感性和特异性,以便更好地满足研究的需求。
2. BLAST参数的分类BLAST参数可以分为两类:搜索参数(search parameters)和输出参数(output parameters)。
2.1 搜索参数搜索参数用于控制BLAST比对的敏感性和速度。
常见的搜索参数包括:•-query:指定查询序列文件或序列字符串,可以是FASTA格式或GenBank 格式。
•-subject:指定被比对序列文件或序列字符串,可以是FASTA格式或GenBank格式。
•-task:指定比对任务的类型,如blastn、blastp、blastx等。
•-evalue:设置期望值(E-value)的阈值,用于筛选显著的比对结果。
•-word_size:设置比对时使用的单词大小,影响比对的敏感性和速度。
•-gapopen:设置开启一个gap的惩罚分数。
•-gapextend:设置扩展一个gap的惩罚分数。
2.2 输出参数输出参数用于控制BLAST比对结果的格式和内容。
常见的输出参数包括:•-outfmt:设置输出格式,如0表示默认格式,5表示XML格式。
•-out:指定比对结果的输出文件。
•-max_target_seqs:设置返回的最大比对序列数目。
•-num_threads:设置线程数,用于加速比对过程。
•-num_alignments:设置返回的最大比对结果数目。
•-max_hsps:设置每个比对结果返回的最大高分片段数目。
3. 如何选择合适的blast参数?选择合适的blast参数是进行BLAST比对的关键步骤,以下是一些选择参数的建议:3.1 根据比对任务类型选择参数不同的比对任务类型需要使用不同的参数。

生物信息学中的序列比对算法及评估指标比较序列比对是生物信息学中非常重要的工具之一,用于分析和比较生物序列的相似性和差异。
序列比对是理解生物进化和功能注释的关键步骤,在基因组学、蛋白质学和遗传学等领域都有广泛应用。
本文将介绍序列比对的算法原理和常用的评估指标,并对几种常见的序列比对算法进行比较。
一、序列比对算法1.全局比对算法全局比对算法用于比较整个序列的相似性,常见的算法有Needleman-Wunsch 算法和Smith-Waterman算法。
这两种算法都是动态规划算法,其中Needleman-Wunsch算法用于比较两个序列的相似性,而Smith-Waterman算法用于寻找局部相似的片段。
这些算法考虑了序列的整体结构,但在处理大规模序列时计算量较大。
2.局部比对算法局部比对算法用于找出两个序列中最相似的片段,常见的算法有BLAST (Basic Local Alignment Search Tool)算法和FASTA(Fast All)算法。
这些算法以快速速度和高敏感性著称,它们将序列切割成小的段落进行比对,并使用统计模型和启发式搜索来快速找到最佳匹配。
3.多序列比对算法多序列比对算法用于比较多个序列的相似性,常见的算法有ClustalW和MAFFT(Multiple Alignment using Fast Fourier Transform)算法。
这些算法通过多次序列比对来找到共有的特征和区域,并生成多序列的一致性描述。
二、评估指标1.一致性分数(Consistency Score)一致性分数是衡量序列比对结果一致性的指标,它反映了序列比对的精确性和准确性。
一致性分数越高,表示比对结果越可靠。
常用的一致性分数有百分比一致性(Percentage Identity)和序列相似度(Sequence Similarity)。
2.延伸性(Extension)延伸性是衡量序列比对结果的长度的指标。

生物信息研究中的序列对齐与比对算法研究序列对齐与比对算法在生物信息研究中扮演着至关重要的角色。
生物信息学是一门研究生物大分子之间的相似性和差异性的学科,它涉及到生命科学、计算机科学和统计学等多个领域的交叉。
序列对齐是生物信息学中的一项基础工作,旨在寻找和比较两个或多个生物序列(如DNA、RNA或蛋白质序列)之间的相似性和差异性。
本文将介绍序列对齐的基本原理、常用算法以及其在生物信息研究中的应用。
首先,我们来解释一下序列对齐的基本概念。
在生物学中,序列是指基因组中的碱基序列或蛋白质中的氨基酸序列。
序列对齐是将两个或多个序列进行比对,并找到它们之间的相似性和差异性的过程。
序列对齐通常分为全局对齐和局部对齐两种类型。
全局对齐旨在比较整个序列,而局部对齐则重点关注序列中的一部分区域。
序列对齐可以揭示生物分子的进化关系、功能预测以及寻找序列中的共同特征。
序列对齐的方法有多种,其中最常用的算法是Smith-Waterman算法和Needleman-Wunsch算法。
Smith-Waterman算法是一种局部序列比对算法,它通过构建一个得分矩阵,并根据得分矩阵找到两个序列中最佳的相似区域。
Needleman-Wunsch算法是一种全局序列比对算法,它通过动态规划的方法,建立一个得分矩阵,并找到两个序列中的最佳匹配。
这些算法都是基于动态规划的思想,通过寻找最优的对齐方案来确定序列的相似性。
除了Smith-Waterman和Needleman-Wunsch算法,还有一些其他的序列比对算法,如BLAST算法和FASTA算法。
BLAST算法是一种常用的快速比对算法,它通过将查询序列与数据库中的序列进行比对,找到最相似的序列。
FASTA算法也是一种常用的快速比对算法,它通过构建一个特殊的索引,加速序列的比对过程。
这些比对算法的不同之处在于其运行速度、准确性和适用范围。
序列对齐和比对算法在生物信息研究中有着广泛的应用。
首先,它们可以用来研究物种的进化关系。

BLAST使用方法一、BLAST的安装和准备工作2.获取待比对的序列文件,可以是FASTA格式的DNA或蛋白质序列。
二、BLAST的常用参数和选项1. Program:指定使用哪种BLAST程序(如BLASTn、BLASTp等)。
2. Database:指定使用哪个数据库进行比对。
3. Query:指定待比对的序列文件。
4. E-value:期望值。
一种描述比对结果误差率的指标,值越小表示结果越可信。
通常情况下,E-value小于0.01被认为是显著结果。
5. Word size:BLAST在比对时使用的核心词的长度。
长度越大表示查全率(sensitivity)越高,但速度会减慢。
6. Gap open:允许在比对过程中插入空位(如插入一个碱基)。
Gap open参数定义了开放一个空位的惩罚分数。
7. Gap extension:允许空位的延伸。
Gap extension参数定义了延伸一个空位的惩罚分数。
三、使用BLAST进行比对1.命令行方式:-打开命令行界面,并定位到BLAST软件的安装目录。
- 输入命令,指定BLAST程序、数据库、查询文件和其他参数。
例如:blastn -db nt -query query.fasta -out output.txt -evalue 0.01-运行命令,BLAST将开始进行比对并生成结果文件。
2.网页方式(以NCBIBLAST为例):- 打开NCBI网站的BLAST页面()。
-选择需要使用的BLAST程序(如BLASTn、BLASTp等)。
-上传待比对的序列文件,或者粘贴序列文本到输入框中。
-选择适当的数据库和其他参数。
-点击“BLAST”按钮,等待比对完成。
四、解读BLAST结果1. E-value:表示在随机比对中获得与查询序列相似度更高的结果的期望概率。
E-value越小表示比对结果越显著。
2. Bitscore:用于表示比对结果的质量。
Bitscore越高表示比对结果越可信。


BLAST 应用专题实践摘要BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。
BLAST中的常用程序有BLASTP(蛋白质序列到蛋白质库的查询)、BLASTX是核酸序列到蛋白库中的一种查询、BLASTN(核酸序列到核酸库中的一种查询)、TBLASTN(蛋白序列到核酸库中的一种查询)、TBLASTX(核酸序列到核酸库中的一种查询)。
BLAST与FASTA是当今最流行的两种比对程序,在生物学中被广泛应用。
本文主要介绍DNA序列比对和蛋白质序列比对。
ABSTRACTBLAST use a partial algorithm to find the similar sequences between two sequences. There are some common used program in the BLAST : BLASTP, BLASTX, BLASTN, TBASTN, TBLASTX. Today, BLAST and FASTA is two of the most popular comparing program, which is widely used in the biology field. This paper mainly introduces the alignment of DNA sequences and protein sequeces.一、前言Blast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
本文通过使用NCBI中的BLAST比对DNA序列和蛋白质序列,重点介绍了BLAST页面中各项参数的功能。
二、DNA序列比对2.1准备工作进入/Blast.cgi ,点击nucleotide blast 出现“Enter Query Sequence”的文本框,输入需要进行比对的序列,如选择:CCTCCCCCTTTGCTTTTTGCTCTCTTGTTAGTATATTAATTGTTTTCACTC TCTGAATCTTTTTTCCCCATTTCTTTGGCAGACATTTTTACTTGTCTTGGAAGAGTAG GTGAAGAGCTGTTTTTAGGACTCTTTGAAAGGGTACAGTATGGGTGACAGTCT>11462Query subrange 可以从需要比对的序列中截取一段,其中from和to代表起始和中指的位置。

实习二BLAST和Fasta的使用
实习目标
1.掌握BLAST的基本功能并可熟练使用
2.了解FastA的基本功能及使用方法
实习内容
1.BLAST
(1).在NCBI中搜索人或动物任一基因的核苷酸序列。
/
(2).用NCBI中的BLAST在线工具搜索与其相匹配的序列。
(3).在EBI中做BLAST找到与所给序列相匹配的序列
/blastall/
*2.Fasta
(1).打开网址/fasta33/进入Fasta 的查询网址(2).浏览并练习Fasta 的查询方法
一个相关的网站:/pages/lab.htm
注:供参考的基因: Myoglobin肌红蛋白,HB血红蛋白,hiv等。
思考题
1.比较两种查询方法的不同与优劣
2.比较在NCBI和EBI中blast查询结果的不同
实习三序列对位显著性检验的计算机模拟
实习目标
1. 通过编写程序更深刻的理解动态规划法的原理步骤
2. 编写显著性检验的程序并输出结果来理解显著性检验的原理
实习内容
1.编写所给序列的局部对位的程序(动态规划法)
2.得出最优分并记录
3.编写显著性检验程序
(1).随机打乱所给两条序列
(2).进行局部对位
(3)反复循环(>10000) 记录结果
4. 求出p-value值并画出得分分布图
注:可能用到的VB语法及函数: 数组, 循环, 判断语句,随机数的产生,文件的读写保存等.
思考题
1.观察随机序列分值的分布情况
2.解释两条序列局部对位分值与相似性的关系。

生物信息学中的序列比对方法序列比对是生物信息学中一项非常重要的工具,其主要目的是将两个或更多的DNA、RNA或蛋白质序列进行比较,以找到它们之间的相似性和差异性。
这样的比对可以用来识别基因、预测蛋白质结构、推断进化关系和研究生物系统的复杂性等。
随着DNA测序技术的快速发展,越来越多的生物学家和生物信息学家开始研究序列比对方法。
序列比对是一项复杂而耗时的任务,需要对大量的序列进行计算和分析。
因此,发展高效的序列比对方法对于生物信息学的发展至关重要。
当前,生物信息学界广泛应用的序列比对方法主要包括全局比对、局部比对和多序列比对等。
一、全局比对全局比对是指将整个序列与另一个相似序列进行比对。
它的应用场景通常是在两个相对较短的序列中查找相似片段,以便在进一步的研究中进行详细的分析。
全局比对方法最常用的是Needleman-Wunsch算法和Smith-Waterman算法。
Needleman-Wunsch(NW)算法是第一个被开发出来的全局比对算法。
该算法基于动态编程的思想,通过将整个序列进行比对,计算出最佳匹配的得分和路径。
然而,这种方法的时间复杂度非常高,随着序列长度的增加,其计算成本也会呈指数级增长。
Smith-Waterman(SW)算法是一种优化的全局比对算法,其核心思想与NW算法类似。
不同之处在于SW算法将匹配的得分设置为正数,而将多余的间隔和未匹配的子序列得分设置为负数。
通过这种方式,SW算法可以得到一个全局最佳的比对结果。
然而,该算法的计算成本也比较高,因此其应用场景受到一定的限制。
二、局部比对局部比对是指在比对序列的过程中,只对部分区域进行比对。
与全局比对不同,局部比对更适用于两个序列之间只有一些片段相似的情况。
常用的局部比对方法主要包括BLAST算法和FASTA算法等。
BLAST算法是一种聚集序列算法,它将大量的搜索序列放入一个空间中,通过加速计算找到最匹配的序列。
通过BLAST算法,可以快速搜索数据库中的所有序列,并找到与目标序列相似的匹配。

多序列比对方法多序列比对是生物信息学中一个常见的分析方法,用于比较多个序列之间的相似性和差异性。
本文将介绍多序列比对的基本原理、常用方法和软件工具,以及其在生物学研究中的应用。
一、多序列比对的基本原理多序列比对是指对多个生物序列进行比较和分析。
生物序列可以是蛋白质序列、DNA序列或RNA序列等。
多序列比对的主要目的是确定序列之间的保守区域和变异区域,并发现序列之间的结构和功能相关性。
多序列比对的基本原理是通过构建序列之间的相似性矩阵,确定最佳的比对结果。
相似性矩阵用于测量两个序列之间的相似性,通常使用BLOSUM、PAM或Dayhoff矩阵等。
基于相似性矩阵和动态规划算法,可以计算序列之间的最佳比对路径,以及比对的得分。
二、常用的多序列比对方法1. 基于全局比对的方法:该方法适用于序列之间的整体相似性比较,常用的算法有Needleman-Wunsch算法和Smith-Waterman算法。
这两种算法都采用动态规划策略,通过计算各种可能的比对路径来确定最佳比对结果。
全局比对方法的主要缺点是在序列相似性较低的情况下,比对结果可能不准确。
2. 基于局部比对的方法:该方法适用于序列之间的部分相似性比较,常用的算法有BLAST和FASTA。
局部比对方法主要通过搜索局部相似片段来进行比对,可以提高比对的敏感性和准确性。
BLAST和FASTA是两种常用的快速局部比对工具,可以快速比对大规模序列数据库。
3. 基于多重比对的方法:该方法适用于多个序列之间的比较和分析,常用的算法有ClustalW和MAFFT。
多重比对方法通过构建多个序列的比对结果,可以识别序列之间的共同保守区域和变异区域,以及序列的结构和功能相关性。
ClustalW和MAFFT是两种常用的多重比对工具,具有较高的准确性和可靠性。
三、常用的多序列比对软件工具1. ClustalW:ClustalW是一个常用的多重比对软件,主要用于比对蛋白质和DNA序列。

生物信息学的基本方法和应用生物信息学是一门近几十年来发展迅速的交叉学科,涉及生物学、物理学、计算机科学、数学等多个领域,其主要任务是利用计算机技术来处理、分析和利用生物信息数据,以解决生物学中的重大问题。
生物信息学常用的工具包括基于序列的分析、基于结构的分析、基于功能的分析和生物网络分析等。
下面我们就来看一下生物信息学的基本方法和应用。
一、基于序列的分析基于序列的分析是生物信息学中最基本的分析方法。
它主要基于DNA、RNA或蛋白质序列的比对和相似性计算来进行。
常见的序列分析工具包括BLAST、FASTA、ClustalW等。
BLAST是目前最常用的序列比对工具之一,它能够通过比对相似序列来推测未知序列的功能。
FASTA和ClustalW也是常用的序列比对工具,它们可以比较多个序列间的相似性,较好地完成序列比对工作。
基于序列的分析可应用于基因注释、基因组比较、系统发育分析等,是生物信息学研究的重要工具。
二、基于结构的分析基于结构的分析主要是通过计算蛋白质的二级结构、三级结构或结合位点等信息进行分析。
通过蛋白质结构的比对和相似性计算可以推测其功能、进行药物研究等。
常见的基于结构的分析工具包括PDB、MolProbity、DOCK等。
PDB是全球公认的蛋白质结构数据库,提供了大量的蛋白质结构信息。
MolProbity可以用于评价蛋白质结构的质量,DOCK则可用于药物分子的分子对接和筛选。
基于结构的分析可以应用于药物设计、酶学研究、基因调控研究等,其研究价值非常高。
三、基于功能的分析基于功能的分析主要是通过对基因、基因产物的功能进行预测和分析。
常见的基于功能的分析工具包括KEGG、GO、DAVID 等。
KEGG是一种常用的基因注释工具,它提供了大量的代谢通路、遗传学和蛋白质家族信息。
GO是一个功能注释数据库,通过对GO注释进行统计分析,可以推测某个基因是否与某个生物过程或功能相关。
DAVID则可以进行大规模基因列表的分析和注释。
生物信息学软件的基本使用方法介绍生物信息学是研究生物学中大规模数据的获取、存储、管理、分析和解释的学科。
为了能够有效地处理这些复杂的生物数据,生物信息学研究者使用了许多专门设计的软件工具。
本文将介绍几种常见的生物信息学软件,并提供基本的使用方法。
1. BLAST(Basic Local Alignment Search Tool):BLAST是一种用于基因序列比对和相似性搜索的软件工具。
它能够找到在数据库中与输入序列相似的序列,并计算它们之间的相似度分数。
使用BLAST时,首先需要选择要比对的数据库,如NCBI的nr数据库。
然后,将待比对的序列输入到BLAST中,并选择合适的算法和参数,最后点击运行按钮即可得到比对结果。
2. ClustalW:ClustalW是一种常用的多序列比对软件。
它能够将多个序列对齐,并生成比对结果。
使用ClustalW 时,首先需要输入要比对的序列。
可以通过手动输入、从文件中导入或从数据库中获取序列。
然后,选择合适的比对算法和参数,并点击运行按钮。
在比对结果中,会显示相似性分数矩阵和序列的对齐信息。
3. FASTA:FASTA是一种用于快速比对和搜索序列相似性的工具。
它使用一种快速的搜索算法,能够在大型数据库中快速找到与输入序列相似的序列。
使用FASTA时,需要将待比对的序列输入到软件中,并选择匹配的算法和搜索参数。
运行后,软件会生成相似序列的列表和相似性评分。
4. R:R是一种统计分析软件,也被广泛用于生物信息学领域。
它提供了丰富的函数和库供生物信息学研究者使用,用于数据处理、统计分析和可视化。
使用R时,可以通过命令行或脚本编写代码来执行各种操作。
例如,可以使用R中的Bioconductor库进行基因表达数据的分析和可视化。
5. IGV(Integrative Genomics Viewer):IGV是一种用于基因组数据可视化的软件工具。
它能够显示基因组位置上的测序深度、SNP、CNV等信息,并支持交互式操作和注释查看。
基因序列分析中的比对算法研究一、引言基因是生命的基本单元,基因在不断的进化和演化中不断累积变异。
对基因序列进行比对分析可以研究基因的演化和遗传多样性等问题。
比对算法是基因序列分析的重要工具之一。
目前,比对算法主要分为全局比对和局部比对两种,全局比对适用于相似序列比较,局部比对适用于较短的序列查询。
本文将围绕基因序列比对算法的分类、算法原理和适用范围进行论述。
二、基因序列比对算法分类1. 全局比对算法全局比对算法是指将基因序列的整体进行比对的算法,常用的算法有双向比对算法和Smith-Waterman算法。
双向比对算法在比对过程中使用两条序列进行互相匹配,以找到共同的序列段。
而Smith-Waterman算法则是在序列相似性分析中应用最广泛的局部比对算法,其特点在于能够准确匹配整个序列。
2. 局部比对算法局部比对算法比全局比对算法更加适用于基因序列中寻找相似的片段,而不需要将所有的序列进行比对。
常用的局部比对算法有BLAST算法和FASTA算法。
在这两个算法中,BLAST算法是更为广泛使用的一种,这种算法使用了一个预处理步骤,即将序列数据转化为能够被快速搜索的形式。
三、算法原理1. 双向比对算法在双向比对算法中,将两个序列的每一个碱基进行比对,若一旦有一个碱基不同,则会停止比对。
该算法的优点是比对速度较快。
该算法有一个明显的副作用,即假定一个序列包含另一个序列的全部内容,并将其定位在某个位置。
但是,在一组输入的数据中,两个包含部分内容的序列可能更加的相似。
因此,双向比对算法将导致相似度被夸大的风险。
2. Smith-Waterman算法Smith-Waterman算法将每个序列中的每个碱基逐一比对,并构建一个得分矩阵,该矩阵将包含每个位置的置信值,而每个置信值将表示的是该位置是否可以匹配另一个序列。
该算法的优点是可以发现相似性更小的序列,其缺点是在处理长序列时需要更多的计算时间和更大的内存使用。
3. BLAST算法BLAST算法是一个广泛使用的局部比对算法,该算法是首先将查询序列拆分成较小的片段,然后对每个片段进行匹配,记录得分最高的匹配结果。
生物信息学中的序列比对算法生物信息学是一门交叉学科,它融合了计算机科学、数学、物理学、化学和生命科学等多个学科。
其中,序列比对算法是生物信息学中的一个重要分支。
序列比对是指在两个序列之间找到相同或相似的部分以及它们的位置,它是了解基因、蛋白质等生物大分子的结构和功能的基础。
序列比对算法通常可分为全局比对和局部比对两类。
全局比对是指将两个序列的整个长度进行比较,如Needleman-Wunsch算法、Smith-Waterman算法等。
而局部比对则是将两个序列的一部分进行比较,如BLAST算法、FASTA算法等。
Needleman-Wunsch算法是一种典型的全局序列比对算法。
其基本思想是将待比较的两个序列分别以行和列的形式写成矩阵,然后通过动态规划的方式来寻找最优比对路径。
在计算比对路径的过程中,会涉及到每个位置上的得分以及得分的计算方法。
矩阵左上角的位置代表两个序列均为空时的得分,而得分的计算则是依据设定的匹配得分、代价得分和惩罚得分来计算。
匹配得分表示两个相同的字符或修饰基间的得分,代价得分表示不同的字符或修饰基间的代价,惩罚得分则表示一个序列在与另一个序列进行比对的过程中,可能存在一个序列的片段与另一个序列完全不匹配的情况。
Smith-Waterman算法是另一种全局序列比对算法。
其基本思想和Needleman-Wunsch算法类似,只是在比对路径的寻找过程中进行了一些优化。
在Smith-Waterman算法中,比对路径是从得分最高的点开始构建的,而在Needleman-Wunsch算法中则是从矩阵的右下角开始构建。
此外,Smith-Waterman算法在计算得分时,会将贡献值小于零的得分设置为0。
这样,当比对的两个序列之间存在相对次优的部分匹配时,Smith-Waterman算法可以将其排除在外,得到最优的比对结果。
BLAST算法和FASTA算法则是两种常见的局部序列比对算法。
这两种算法都采用了启发式方法,即通过一系列的筛选步骤来减少不必要的计算,提高比对速度。
生物信息学中的基因组序列比对算法分析在生物信息学研究中,基因组序列比对算法是一项关键技术,它用于比较不同物种或个体的基因组序列,以揭示它们之间的相似性和差异性。
这些算法对于理解生物进化、基因功能和遗传变异等方面至关重要。
本文将介绍几种常见的基因组序列比对算法,并分析其优缺点及适用范围。
1. 简介基因组序列比对是将一个序列与一个参考序列进行比较,找出它们之间的相同或相似的部分。
这种比对有助于研究物种在进化过程中的关系,揭示基因之间的同源性和功能以及识别突变位点等。
基因组序列比对算法分为全局比对和局部比对两类。
2. 全局比对算法全局比对算法旨在找到两个序列之间的最佳匹配,通常使用动态规划方法,最常见的全局比对算法是古典的Needleman-Wunsch算法。
Needleman-Wunsch算法将两个序列表示为一个二维矩阵,然后通过填充矩阵中的格点来计算匹配得分。
该算法考虑了所有可能的比对方式,并且能够找到最佳的匹配方案。
然而,由于需要计算整个序列的所有可能对,该算法的时间复杂度较高,不适用于大规模基因组序列的比对。
3. 局部比对算法局部比对算法是为了找到两个序列中的局部相似部分。
Smith-Waterman算法是最常见的局部比对算法之一。
Smith-Waterman算法与Needleman-Wunsch算法相似,但它在计算匹配分数时,忽略了负分数。
该算法将负分数替换为零,可以找到序列中的局部相似片段,而不仅仅是最佳匹配。
这使得它在识别突变和插入/删除等局部变异时更加灵活。
4. 近似比对算法对于大规模基因组序列的比对,全局和局部比对算法效率较低。
近似比对算法被引入用于加速大规模基因组序列的比对。
经典的近似比对算法包括BLAST和FASTA。
BLAST算法采用一种先搜索数据库中短序列片段的策略,利用预先计算出的索引表来加速搜索过程。
它根据核苷酸或氨基酸的局部片段来找到相似的序列,因此不是全局比对算法,但它速度非常快。
实习三相似序列的数据库搜索 BLAST/FASTA一、实习目的掌握BLAST和FASTA数据检索方法。
二、实习内容BLAST和FASTA程序是目前最常用的基于局部相似性的数据库搜索程序,它们都基于查找完全匹配的短小序列片段,并将它们延伸得到较长的相似性匹配。
它们的优势在于可以在普通的计算机系统上运行,而不必依赖计算机硬件系统而解决运行速度问题。
(一)BLASTBLAST (Basic Local Alignment Search Tool)是NCBI提供的进行序列相似性搜索的工具。
BLAST算法得基本思路是首先找出检测序列和目标序列之间相似性程度最高得片段,并作为内核向两端延伸,以找出尽可能长得相似序列片段。
BLAST程序之所以使用广泛,主要因为其运行速度比FASTA等其它数据库搜索软件要快。
与ENTREZ提供的文本搜索不同,BLAST以核酸或蛋白质序列作为搜索条件(query),搜索指定数据库中与query相似度较高,甚至同源的序列。
/Blast.cgi(二)BLASTPPSI-Blast和PHI-Blast是在普通BLAST的基础上发展起来的两个新程序,是将双序列比对和多序列比对结合在一起的数据库搜索方法,可通过多次迭代搜索出蛋白质家族或超家族中序列相似性较低的成员,弥补了普通BLAST难于找到进化距离较远的同源序列的不足。
PSI-Blast程序的主要思想是通过多次迭代找出最佳结果。
第一次blast搜索后,利用结果中最相似的序列重新构建位点特异性打分矩阵(PSSM),然后再使用该矩阵进行第二轮blast搜索,再调整矩阵,搜索,如此迭代,直到找出最佳搜索结果。
最终高度保守的区域就会得到比较高的分值,而不保守的区域则分数降低。
这样可以提高blast搜索的灵敏度,从而有利于发现进化距离较为遥远的同源序列。
三、作业在NCBI上搜索拟南芥(Arabidopsis thaliana)的A TP sulfurylase的核酸和蛋白质序列,用它尝试进行BLASTN和PSI-BLAST搜索,给出每一步搜索的参数设置及选择原因,分析搜索结果(找到的是其它物种中的ATP sulfurylase,还是执行其它相似功能的序列)。
在线b l a s t的用法总结-标准化文件发布号:(9556-EUATWK-MWUB-WUNN-INNUL-DDQTY-KIIBlast(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST 采用一种局部的算法获得两个序列中具有相似性的序列NCBI的在线blast:/Blast.cgi本文详细出处参考:/475/举例一:核酸序列的比对1,进入在线blast界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。
不同的blast程序上面已经有了介绍。
这里以常用的核酸库作为例子。
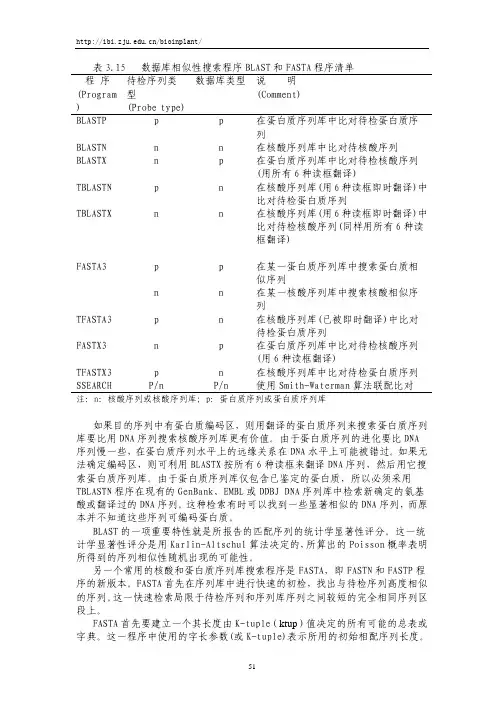
(补充介绍下:1、BLASTN【 nucleotide blast】是核酸序列到核酸库中的一种查询。
库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
2、BLASTP【protein blast】是蛋白序列到蛋白库中的一种查询。
库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
3、BLASTX是核酸序列到蛋白库中的一种查询。
先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
4、TBLASTN是蛋白序列到核酸库中的一种查询。
与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
5、TBLASTX是核酸序列到核酸库中的一种查询。
此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
)2,粘贴fasta格式的序列。
选择一个要比对的数据库。
关于数据库的说明请看NCBI在线blast数据库的简要说明。
一般的话参数默认。
3,blast参数的设置。