总胆汁酸测定试剂盒(酶循环法)产品技术要求九强
- 格式:docx
- 大小:28.35 KB
- 文档页数:4

总胆汁酸测定试剂盒(酶循环法)适用范围:本试剂盒用于体外定量测定人血清中总胆汁酸的浓度。
1.1包装规格试剂1:1×30ml;试剂2:1×10ml试剂1:2×30ml;试剂2:1×20ml试剂1:1×60ml;试剂2:1×20ml试剂1:3×80ml;试剂2:4×20ml试剂1:4×60ml;试剂2:4×20ml试剂1:2×60ml;试剂2:2×20ml试剂1:2×30ml;试剂2:2×10ml试剂1:6×60ml;试剂2:2×60ml2.1 外观试剂1为无色至黄色液体,试剂2为无色至浅黄色液体,无可见异物。
2.2 净含量试剂的净含量不少于标称装量。
2.3 试剂空白2.3.1 试剂空白吸光度在37℃、波长405nm、1.0cm光径条件下,用生理盐水作为样本加入试剂测试时,试剂空白吸光度应不大于0.80。
2.3.2 试剂空白吸光度变化率在37℃、波长405nm、1.0cm光径条件下,用生理盐水作为样本加入试剂测试时,试剂空白吸光度的变化应不大于0.04/min。
2.4 分析灵敏度TBA含量为50umol/L时,测定吸光度变化率>0.020△A/min。
2.5 线性区间试剂(盒)线性在[1.0,150]umol/L区间内:2.5.1 线性相关系数(r)应不小于0.9900;2.5.2 [1.0,20]umol/L区间内,线性绝对偏差不超过±4umol/L;(20,150]umol/L区间内,线性相对偏差不超过±10%。
2.6 精密度2.6.1 重复性用相同批号试剂盒测试两个水平的质控品,批内变异系数(CV)应不大于5.0%。
2.6.2 批间差用3个不同批号试剂盒测试两个水平的质控品,批间相对极差应不大于10.0%。
2.7 准确度与已上市的同类产品比对,用40个在[1.0,150]umol/L区间内不同浓度的人源样本,用线性回归方法计算两组结果的相关系数(r)不小于0.9900;[1.0,20]umol/L区间内,线性绝对偏差不超过±4umol/L;(20,150]umol/L区间内,线性相对偏差不超过±10%。

总胆汁酸测定试剂盒(酶循环法)适用范围:本试剂用于体外定量测定人体血清中总胆汁酸的含量。
1.1 包装规格a) 试剂1:2×45 mL,试剂2:2×15 mL;b) 试剂1:4×60 mL,试剂2:4×20mL;c) 试剂1:2×60mL,试剂2:2×20mL。
1.2主要组成成分1.2.1试剂1主要组分磷酸氢二钠-柠檬酸缓冲液100mmol/L氧化型硫代辅酶(THIO-NAD) 950mg/L还原型辅酶(NADH) 6g/L聚氧乙烯油醇醚适量1.2.2试剂2主要组分Tris缓冲液100mmol/L3α-羟基类固醇脱氢酶(3α-HSDH) 12.5KU/L聚氧乙烯油醇醚适量2.1外观试剂1:浅黄绿色澄清液体;试剂2:无色澄清液体。
2.2试剂装量应不低于试剂瓶标示装量。
2.3 试剂空白2.3.1试剂空白吸光度:在405nm处测定试剂空白吸光度,应≤0.8。
2.3.2试剂空白吸光度变化率:在405nm处测定空白吸光度变化率|△A/min|≤0.05。
2.4分析灵敏度测定TBA含量为10 µmol/L样本时,其|△A/min|应≥0.01。
2.5线性范围2.5.1测试浓度在[0,180] µmol/LL范围内,线性回归的相关系数(r)应不低于0.990;2.5.2测试浓度在[0,20] µmol/L范围内,线性绝对偏差应不超过±2µmol/L;测试浓度在(20,180] µmol/L范围内,线性相对偏差应不超过±10%。
2.6 测量精密度2.6.1重复性:重复测试三个水平的样本,所得结果的变异系数(CV)应不大于5%。
2.6.2批间差:抽取3个不同批号的试剂,对同一份样本进行重复测定,相对极差≤10%。
2.7准确度比对试验:相关系数r≥0.975,各个浓度点中≤20 µmol /L的绝对偏差不超过±2µmol /L。

性能指标
2.1外观
试剂盒中液体试剂外观应澄清、透明,无沉淀、悬浮物和絮状物。
2.2净含量
液体试剂的净含量应不少于标示值。
2.3试剂空白
试剂盒以纯化水为空白在37℃ 1℃,405nm主波长条件下,试剂空白吸光度应≤1.000,试剂空白吸光度变化率(△A/min)应≤0.005。
2.4分析灵敏度
试剂盒测定浓度为50.0µmol/L的样品时,吸光度变化率(△A/min)应≥0.010。
2.5精密度
2.5.1试剂盒的批内不精密度变异系数(CV)应≤
3.00%。
2.5.2试剂盒的批间不精密度相对极差(R)应≤ 4.00%。
2.6准确度
试剂盒的准确度相对偏差应在±10.0%范围内。
2.7线性范围
试剂盒在2.0μmol/L~180.0μmol/L的范围内,其线性相关系数(r)应≥0.9900,线性相对偏差应在±10.0%以内。

总胆汁酸测定试剂盒(酶循环法)适用范围:用于定量检测人血清中总胆汁酸(TBA)浓度。
1.1包装规格a) 试剂1:2×45ml,试剂2:2×15ml;b) 试剂1:4×45ml,试剂2:4×15ml;c) 试剂1:6×60ml,试剂2:2×60ml;d)试剂1:3×60ml,试剂2:3×20ml;e)试剂1:2×300ml,试剂2:2×100ml;f)试剂1:12×16.8ml,试剂2:12×5.6ml;g)试剂1:2×54ml,试剂2:2×18ml;h)试剂1:1×30ml,试剂2:1×10ml。
1.2试剂主要组成成分试剂1主要组成成分甘氨酸缓冲液25 mmol/Lβ-硫烟酰胺腺嘌呤二核苷氧化型(Thio-NAD) 1 mmol/L试剂2主要组成成分Tris缓冲液25 mmol/L烟酰胺腺嘌呤二核苷还原型(NADH)10 mmol/L3α-羟类固醇脱氢酶(3α-HSD)12500 U/L2.1 外观和性状2.1.1 试剂盒各组分应齐全、完整、液体无渗漏;外包装完好、无破损,标签完好、字迹清晰。
2.1.2 试剂1应为淡黄色透明溶液;试剂2应为无色或淡黄色透明溶液。
2.2 净含量应不低于试剂瓶标示装量。
2.3 试剂空白2.3.1 试剂空白吸光度测定试剂空白吸光度,应<0.5。
2.3.2 试剂空白吸光度变化率试剂空白吸光度变化率△A/min≤0.04。
2.4 分析灵敏度测试150umol/L的被测物时,吸光度变化率(ΔA/min)应不低于0.02。
2.5 准确度回收率90%~110% 范围内。
2.6 精密度2.6.1批内精密度批内变异系数应不大于5%。
2.6.2批间精密度批间相对极差应不大于10%。
2.7 线性区间2.7.1在[2,180)umol/L区间内,线性回归的相关系数(r)应不低于0.990;2.7.2在(50,180)umol/L区间内,相对偏差不超过±15%;2.7.3在[2,50]umol/L区间内,绝对偏差不超过±5umol/L。

总胆汁酸测定试剂盒(酶循环法)适用范围:本试剂盒与ABBOTT ARCHITECT c4000/c8000/c16000全自动生化分析仪配套使用,用于体外定量测定人血清中总胆汁酸的浓度。
1.1包装规格液体双剂型试剂1(R1):60mL×2,试剂2(R2):20mL×2,校准品:2mL×1;试剂1(R1):53mL×2,试剂2(R2):20mL×2,校准品:2mL×1;试剂1(R1):70mL×4,试剂2(R2):25mL×4,校准品:2mL×1。
1.2主要组成成分1.2.1 试剂1(R1)(液体)Thio-NAD+952.9mg/L1.2.2 试剂2(R2)(液体)NADH6.1g/L3-αHSD12500U/L 1.2.3 校准品(液体)在Mes-HCL缓冲液中添加甘氨胆酸钠,目标浓度:50.0μmol/L。
(每批定值,值有批特异性,详见值单)2.1 外观试剂盒中各组件的外观应满足:2.1.1 试剂1(R1)应为浅黄色透明溶液,无杂质、无絮状物,外包装完整无破损;2.1.2 试剂2(R2)应为无色透明溶液,无杂质、无絮状物,外包装完整无破损;2.1.3 校准品应为无色透明溶液,无杂质、无絮状物,外包装完整无破损。
2.2 净含量液体试剂净含量应不少于标示值。
2.3 试剂空白吸光度在波长405nm(400nm~420nm)处(光径1cm),试剂空白吸光度(A)应≤0.500;试剂空白吸光度变化率(△A/min)≤0.020。
2.4 准确度用中生试剂和已上市同类试剂分别测定40个在线性范围内不同浓度的样本,在[0.0,180.0]μmol/L检测范围内,比对两组数据的相关系数(r)及测值的偏差,要求r≥0.9900;在(10.0,180.0]μmol/L区间内,相对偏差应不超过±15%;在[0.0,10.0]μmol/L区间内,绝对偏差应不超过±1.5μmol/L。

总胆汁酸(TBA)测定试剂盒
(酶循环法)
2.1外观和性状
外观和性状应符合表2要求。
表2 试剂盒内各组分的外观性状
2.2试剂空白
2.2.1试剂空白吸光度
试剂以蒸馏水为空白时,在波长 405nm,光径 1.0 cm,温度 37℃条件下,吸光度≤0.80。
2.2.2试剂空白吸光度变化率
试剂以蒸馏水为空白时,在波长 405nm,光径 1.0 cm,温度 37℃条件下,吸光度变化率≤0.02。
2.3分析灵敏度
试剂盒测试浓度为10 μmol/L被测物时,吸光度变化率≥0.005。
2.4线性范围
2.4.1试剂盒在2~180 μmol /L区间(范围)内,其回归系数r≥0.9900。
2.4.2相对偏差或绝对偏差应符合表 3 要求。
表3 相对偏差或绝对偏差
2.5精密度
2.5.1试剂盒批内精密度 CV 值应≤5.0%。
2.5.2试剂盒批间相对极差(R)应≤10.0%。
2.6准确度
相对偏差(Bias%)应在参考物质靶值±10%以内。
2.7液体装量
试剂盒不同规格的净含量应不少于其标示量。

总胆汁酸测定试剂盒(酶循环法)适用范围:本试剂用于体外定量测定人血清中总胆汁酸的浓度。
1.1规格液体双剂型试剂1(R1):60mL×2,试剂2(R2):20mL×2,校准品:2mL×1;试剂1(R1):45mL×2,试剂2(R2):15mL×2,校准品:2mL×1。
1.2规格划分说明根据净含量划分规格。
1.3主要组成成分试剂盒由试剂1(R1)液体、试剂2(R2)液体及校准品液体组成。
1.3.1 试剂1(R1)液体:Thio-NAD+ 952.9mg/L1.3.2 试剂2(R2)液体:NADH 6.1g/L3-α HSD 12500U/L1.3.3 校准品液体:Mes-HCL缓冲液为基质甘氨胆酸钠42.5μmol/L ~57.5μmol/L(每批定值)2.1 外观试剂盒中各组件的外观应满足:a) 试剂1(R1)应为浅黄色透明溶液,无杂质、无絮状物,外包装完整无破损;b) 试剂2(R2)应为无色透明溶液,无杂质、无絮状物,外包装完整无破损;c) 校准品应为无色透明溶液,无杂质、无絮状物,外包装完整无破损。
2.2 净含量液体试剂净含量应不少于标示值。
2.3 试剂空白吸光度在波长405nm(400nm~420nm)处(光径1cm),试剂空白吸光度(A)应≤0.500;试剂空白吸光度变化率(△A/min)≤0.020。
2.4 准确度用中生试剂和已上市同类试剂分别测定40个在测定范围内不同浓度的样本,在[0.0,180.0]μmol/L检测范围内,比对两组数据的相关系数(r)及测值的偏差,要求r≥0.9900;在(10.0,180.0]μmol/L区间内,相对偏差应不超过±15%;在[0.0,10.0]μmol/L区间内,绝对偏差应不超过±1.5μmol/L。
2.5 分析灵敏度对应于浓度为50.0μmol/L的TBA所引起的吸光度变化率(△A/min)应在0.010~0.150的范围内。

总胆汁酸测定试剂盒(酶循环法)适用范围:本产品用于体外定量测定人血清中总胆汁酸的含量。
1.1规格具体产品规格见下表:1.2 组成成分试剂1:β-硫代烟酰胺脲嘌呤二核苷酸氧化型(Thio-NAD) 1.0g/L试剂2:3α-羟基类固醇脱氢酶(3α-HSD ) 12.5KU/LNADH6.1g/L2.1 外观2.1.1 外包装完整无破损;2.1.2 试剂1:黄色澄清透明无杂质液体;2.1.3 试剂2:无色澄清透明无杂质液体。
2.2 净含量净含量不低于标示值。
2.3 试剂空白2.3.1 试剂空白吸光度在主波长405nm、副波长660nm、37℃条件下,试剂空白吸光度不大于0.8。
2.3.2 试剂空白吸光度变化率在主波长405nm、副波长660nm、37℃条件下,试剂空白变化率不大于0.04。
2.4 线性2.4.1 线性范围[5.0,150.0]μmol/L,相关系数r不小于0.990。
2.4.2 线性偏差(20.0,150.0]μmol/L线性范围内,相对偏差不超过±15%;[5.0,20.0]μmol/L线性范围内,绝对偏差不超过±4.0μmol/L。
2.5 分析灵敏度检测浓度为26.8μmol/L的样本时,吸光度变化率不小于0.013。
2.6 重复性测试高、低值质控品,重复测试至少10次,CV≤5%。
2.7 批间差用三个不同批号的试剂测试同一样本,重复测试3次,相对极差R≤10%。
2.8 准确度回收率在90%~110%范围内。
2.9 稳定性原包装试剂2~8℃避光储存,有效期12个月。
效期后1个月内产品应符合2.1、2.3、2.4、2.5、2.6和2.8的要求。

总胆汁酸(TBA)测定试剂盒(酶循环法)说明书【产品名称】总胆汁酸(TBA)测定试剂盒(酶循环法)【包装规格】a)试剂1:2×45mL 试剂2:2×15mL b)试剂1:4×60mL 试剂2:4×20mL c)试剂1:2×60mL试剂2:2×20mL【预期用途】用于体外定量测定人体血清中总胆汁酸的含量。
血清中TBA 水平升高见于急性病毒性肝炎、肝硬化、胆汁淤积性胆管炎、慢性活动性肝炎、酒精性肝炎、阻塞性黄疸、肝囊性纤维变性[1]。
【检验原理】胆汁酸被3α-HSD 及THIO-NAD 特异性的氧化,生成3-酮类固及还原型硫代辅酶Ⅰ(THIO -NADH )。
此外,生成的3-酮类固醇在3α羟基类固醇脱氢酶及还原型硫代辅酶Ⅰ(THIO -NADH )存在下,生成胆汁酸及氧化型硫代辅酶Ⅰ(THIO -NAD )。
如上述,据循环酶而放大微量的胆汁酸量,测定在单位时间内生成的还原型硫代辅酶Ⅰ在405nm 处的吸光度变化,可求得样本中总胆汁酸的含量。
【主要组成成分】试剂1主要组分磷酸氢二钠-柠檬酸缓冲液100mmol/L 氧化型硫代辅酶Ⅰ(THIO-NAD)950mg/L还原型辅酶(NADH)6g/L 聚氧乙烯油醇醚适量试剂2主要组分Tris 缓冲液100mmol/L 3α-羟基类固醇脱氢酶(3α-HSDH)12.5KU/L聚氧乙烯油醇醚适量注:不同批号试剂盒中各组分未经试验不可互换。
【储存条件及有效期】贮存于2~8℃,有效期为18个月,生产日期、有效期见标签。
【适用仪器】艾威德AS-420/AS-660/AS-1200;日立HITACHI 7020型/7060型/7180型/7600型/LABOSPECT 008AS 型;贝克曼AU400/AU480/AU640/AU680/AU2700/AU5400/AU5800/AU5811/AU5821;佳能TBA-FX8/TBA-120FR /TBA-2000FR ;罗氏cobas 8000c 702/cobas 8000c 701/cobas 8000c 502;西门子SIEMENS ADVIA 1800/ADVIA 2400;雅培ABBOTT ARCHITECT c8000/ARCHITECT c16000/ARCHITECT ci8200;西森美康SYSMEX BM6010/C ;科华KHB 卓越310/卓越330/卓越400/卓越450/ZY-1200/ZY-1280;迪瑞CS-240/CS-T300/CS-300B/CS-380/CS-400A/CS-400B/CS-600A/CS-600B/CS-800A/CS-800B/CS-1200/CS-1200ISE/CS-1300B/CS-1400;迈瑞MINDRAY BS-220/BS-330/BS-350E/BS-380/BS-390/BS-400/BS-430/BS-600/BS-800/BS-2000M ;颐兰贝ES-200/ES-380/ES-480;赛诺迈德SUNMATIK-9050型;雷杜Chemray 420;英诺华D280;特康TC6010L ;锦瑞GS400;普康6066。
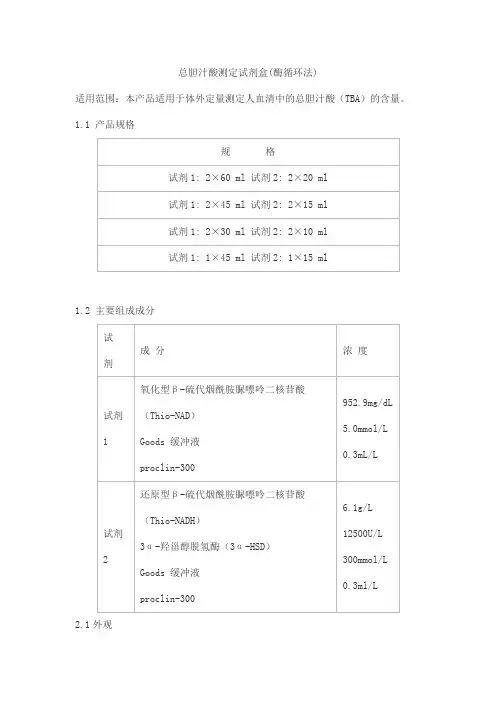
总胆汁酸测定试剂盒(酶循环法)
适用范围:本产品适用于体外定量测定人血清中的总胆汁酸(TBA)的含量。
1.1 产品规格
1.2 主要组成成分
2.1外观
2.1.1试剂盒标签标识清晰,外包装完整无破损;
2.1.2试剂1为黄色澄清液体,目测不得有任何沉淀及絮状悬浮物;
2.1.3试剂2为无色或淡黄色澄清液体,目测不得有任何沉淀及絮状悬浮物。
2.2净含量
净含量不低于标示值。
2.3试剂空白
2.3.1空白吸光度
在405nm(主)、660nm(副)下测定空白吸光度应不大于0.8。
2.3.2空白吸光度变化率
空白变化率△A/min≤0.04。
2.4线性范围
(5,150)μmol/L范围内,相关系数r≥0.990。
2.5分析灵敏度
在产品说明书规定参数设定条件下,测定浓度为40μmol/L样本,△A/min ≥0.020。
2.6 精密度
2.6.1批内重复性
CV≤5.0%。
2.6.2 批间差
相对极差R≤10.0%。
2.7 准确度
与已上市产品比对:(5,150)μmol/L范围内,相关系数r≥0.990;(5,30]μmol/L范围内,绝对偏差不超过±3μmol/L;(30,150)μmol/L范围内,相对偏差不超过±10%。
2.8 稳定性
未开封试剂2℃~8℃储存,可稳定12个月。
取到效期后2个月内产品进行检测,检测结果应满足2.1、2.3、2.4、2.5、2.6.1、2.7的规定。

总胆汁酸测定试剂盒(酶循环法)适用范围:本试剂适用于体外定量测定人血清或血浆中总胆汁酸的含量。
1.1规格R1:4×70ml R2:1×70mlR1:4×50ml R2:2×25mlR1:2×64ml R2:2×16mlR1:2×80ml R2:1×40mlR1:4×45ml R2:1×45ml2.1外观2.1.1包装完整,标签清晰;2.1.2试剂1为无色透明溶液、无悬浮物、无沉淀物;试剂2为淡黄色透明溶液、无悬浮物、无沉淀物;2.2净含量试剂盒内液体的装量应不低于标示值。
2.3试剂空白吸光度用试剂盒测定空白样本,记录试剂盒在505nm 波长、1cm光径条件下,试剂空白吸光度A应不大于0.050。
2.4分析灵敏度测定50.6μmol/L样本时,吸光度变化率(△A/min)应符合:0.0180~0.5000。
2.5线性范围试剂盒线性范围在[0.5,200.0]μmol/L:线性相关系数r≥0.990;[0.5,20.0]μmol/L范围内,线性绝对偏差应不超过±2.0 μmol/L;(20.0,200.0]μmol/L范围内,线性相对偏差不超过±10%。
2.6测量精密度2.6.1 重复性用高、低2个水平的血清样品或质控品测试同一批号试剂盒,测试结果应符合CV≤5%。
2.6.2 批间差用3个不同批号的试剂盒测定同一份样本,测得结果极差R≤10%。
2.7准确度以选定的上市分析系统对照试剂作为比对方法进行方法学比对测试,比对结果应满足:a)在[0.5,200.0]μmol/L范围内,线性相关系数r≥0.990;b)[0.5,20.0]μmol/L范围内,绝对偏差应不超过±2.0 μmol/L;(20.0,200.0]μmol/L范围内,相对偏差不超过±10%。
2.8稳定性试剂盒在2℃-8℃保存条件下贮存到12个月效期后,效期后一个月内进行检测,检测结果应符合2.1、2.3、2.4、2.5、2.6.1、2.7 技术指标要求。
总胆汁酸测定试剂盒(酶循环法)适用范围:用于体外定量测定人血清总胆汁酸的含量。
1.1 包装规格试剂1:63mL×7、试剂2:49mL×3;试剂1:60mL×3、试剂2:60mL×1;试剂1:45mL×4、试剂2:30mL×2;试剂1:60mL×1、试剂2:20mL×1;试剂1:60mL×3、试剂2:20mL×3;试剂1:15mL×1、试剂2:5mL×1;1300测试/盒(试剂1:69mL×2、试剂2:23mL×2);2600测试/盒(试剂1:69mL×4、试剂2:23mL×4);840测试/盒(试剂1:22mL×4、试剂2:15mL×2);840测试/盒(试剂1:45mL×2、试剂2:15mL×2);1350测试/盒(试剂1:48mL×3、试剂2:48mL×1);950测试/盒(试剂1:24mL×2、试剂2:8mL×2);1700测试/盒(试剂1:90mL×2、试剂2:30mL×2);1000测试/盒(试剂1:60mL×3、试剂2:19mL×3);1650测试/盒(试剂1:59mL×3、试剂2:59mL×1);800测试/盒(试剂1:60mL×1、试剂2:19mL×1);2240测试/盒(试剂1:75mL×4、试剂2:29mL×4)。
1.2 组成成分试剂1:磷酸缓冲液适量β-烟酰胺腺嘌呤二核苷酸氧化型950mg/L试剂2:β-烟酰胺腺嘌呤二核苷酸还原型6g/L3α-羟类固醇脱氢酶12.5kU/L2.1 试剂装量应不低于瓶签标示装量。
2.2 外观试剂1:亮黄色澄清液体;试剂2:无色或淡黄色澄清液体。
总胆汁酸测定试剂盒(循环酶法)适用范围:用于体外定量检测人血清中总胆汁酸的浓度。
1.1规格a) 试剂1:2×45ml,试剂2:2×15ml;b) 试剂1:3×60ml,试剂2:3×20ml;c) 试剂1:4×45ml,试剂2:4×15ml;d) 试剂1:2×54ml,试剂2:2×18ml;e) 试剂1:12×16.8ml,试剂2:12×5.6ml;f) 试剂1:2×300ml,试剂2:2×100ml;g) 试剂1:6×60ml,试剂2:2×60ml;h) 试剂1:1×30ml,试剂2:1×10ml;i) 试剂1:2×30ml,试剂2:2×10ml;j) 试剂1:4×60ml,试剂2:4×20ml;k) 试剂1:6×16.8ml,试剂2:6×5.6ml;l) 试剂1:1×45ml,试剂2:1×15ml。
1.2 组成试剂主要组分见表1:表1 试剂主要组分2.1 外观外包装完整无破损,标签清晰;试剂1应为淡黄色透明溶液,试剂2应为无色或淡黄色透明溶液。
2.2 净含量应不低于试剂瓶标示装量。
2.3 试剂空白2.3.1 试剂空白吸光度在405nm处测定试剂空白吸光度,应<0.5;2.3.2 试剂空白吸光度变化率试剂空白吸光度变化率(△A/min)应≤0.04。
2.4 分析灵敏度测定浓度为150μmol/L的样品,吸光度变化率(△A/min)应不低于0.02。
2.5 线性2.5.1在[2,180]μmol/L范围内,线性回归的相关系数应不低于0.990;2.5.2测试浓度[50,180]μmol/L的样品,相对偏差应不超过±10%;测试浓度[2,50)μmol/L的样品,绝对偏差应不超过±5μmol/L。
总胆汁酸(TBA)测定试剂盒(酶循环法)适用范围:本产品用于体外定量测定人血清或血浆中总胆汁酸含量。
1.1规格试剂1(R1): 2×60mL、试剂2(R2): 2×20mL;试剂1(R1): 1×60mL、试剂2(R2): 1×20mL;试剂1(R1): 2×300mL、试剂2(R2): 1×200mL;试剂1(R1): 2×45mL、试剂2(R2): 2×15mL;试剂1(R1): 1×300mL、试剂2(R2): 1×100mL;试剂1(R1): 2×60mL、试剂2(R2): 2×20mL;试剂1(R1): 1×18mL、试剂2(R2): 1×6mL;试剂1(R1):2×75mL、试剂2(R2): 2×25mL;试剂1(R1):1×75mL、试剂2(R2): 1×25mL;试剂1(R1):2×12mL、试剂2(R2): 2×4mL;试剂1(R1):5×12mL、试剂2(R2): 5×4mL;试剂1(R1):10×12mL、试剂2(R2): 10×4mL;试剂1(R1):20×12mL、试剂2(R2): 20×4mL;试剂1(R1):2×15mL、试剂2(R2): 2×5mL;试剂1(R1):5×15mL、试剂2(R2): 5×5mL;试剂1(R1):10×15mL、试剂2(R2): 10×5mL;试剂1(R1):20×15mL、试剂2(R2): 20×5mL;480测试/盒:【试剂1(R1):142.4mL、试剂2(R2):51.2mL】;校准品(选配):1×1mL。
总胆汁酸(TBA)测定试剂盒(酶循环法)适用范围:该试剂盒用于体外定量测定人血清中总胆汁酸的浓度。
1.1 产品规格1.2 组成成分该试剂盒由试剂1(R1)、试剂2(R2)和校准品(选配)组成。
1.2.1试剂组成试剂1: 2-(N-吗啉代)乙磺酸(MES)缓冲液≥20.0mmol/L氧化型硫代烟酰胺腺嘌呤二核苷酸(Thio-NAD+)≥1.0mmol/L试剂2: 三羟甲基甲胺基丙磺酸(TAPS)缓冲液≥100.0mmol/L3-α羟基类固醇脱氢酶(3α-HSD)≥10.0KU/L烟酰胺腺嘌呤二核苷酸(NADH)≥2.0mmol/L1.2.2 校准品组成甘氨脱氧胆酸钠目标浓度:50μmol/L 该校准品为水基质液体校准品2.1 外观a) R1应为淡黄色至黄色溶液,无混浊,无未溶解物。
b) R2应为无色至棕褐色溶液,无混浊,无未溶解物。
c) 校准品应为无色溶液,无混浊,无未溶解物2.2 净含量液体组分不少于标示值。
2.3 试剂空白2.3.1试剂空白吸光度应不大于0.800。
2.3.2试剂空白吸光度变化率应不大于0.04/min。
2.4 分析灵敏度TBA试剂盒测定浓度100μmol/L的被测物时,吸光度差值(ΔA)应不小于0.010。
2.5 准确度回收实验:测定回收率,应在90%-110%之间。
2.6 精密度2.6.1重复性变异系数应不大于5%。
2.6.2批间差批间相对极差(R)应不大于10%。
2.7 线性在(0,180]μmol/L范围内,TBA试剂盒的线性相关系数r应不低于0.9900;在(0,50]范围内绝对偏差应不超过5μmol/L,在(50,180]范围内相对偏差应不超过±10%。
2.8校准品溯源性依据GB/T21415-2008《体外诊断医疗器械生物样品中量的测量校准品控制物质赋值的计量学溯源性》及有关规定提供总胆汁酸校准品的来源、赋值过程以及测量不确定度等内容。
校准品溯源至纯品(Sigma公司,纯度≥97%)。
总胆汁酸测定试剂盒(酶循环法)注册技术审查指导原则(征求意见稿)本指导原则旨在指导注册申请人对总胆汁酸(Total bileacids ,TBA)测定试剂(盒)注册申报资料的准备及撰写,同时也为技术审评部门审评注册申报资料提供参考。
本指导原则是对总胆汁酸测定试剂(盒)的一般要求,申请人应依据产品的具体特性确定其中的具体内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特征对注册申报资料的内容进行充实和细化。
本指导原则是供申请人和审查人员适用的指导文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但应提供详细的研究资料和验证资料。
应在遵循相关法规、国家标准、行业标准的前提下使用本指导原则。
本指导原则是在现行法规、标准体系及当前认知水平下制定的,随着法规、标准体系的不断完善和科学技术的不断发展,本指导原则相关内容也将适时进行调整。
一、适用范围本指导原则适用于基于酶循环法原理,利用全自动生化分析仪、半自动生化分析仪或分光光度计,体外定量测定人血清或血浆中总胆汁酸含量的测定试剂盒。
本指导原则适用于进行首次注册申报和相关许可事项变更的产品。
从方法学考虑,本指导原则适用于采用3α-羟基类固醇脱氢酶(31α-HSD )催化的酶促循环反应,利用全自动、半自动生化分析仪或分光光度计,在405nm 波长附近测定生成的硫代还原型烟酰胺腺嘌呤二核苷酸(Thio-NADH) 的吸光度变化,计算总胆汁酸含量的测定试剂(盒)。
反应原理如图所示。
胆汁酸(Bile acid )被3α- 羟基类固醇脱氢酶 (3 α-HSD )及硫代氧化型烟酰胺腺嘌呤二核苷酸( Thio-NAD +)特异性地氧化,生成3α-酮类固醇( 3α-KS )及硫代还原型烟酰胺腺嘌呤二核苷酸(Thio-NADH) 。
此外,生成的3α- 酮类固醇在3α- 羟基类固醇脱氢酶( 3 α-HSD )及还原型辅酶I (NADH )存在下,生成胆汁酸和氧化型辅酶I(NAD + )。
总胆汁酸测定试剂盒(酶循环法)
适用范围:本试剂盒用于体外定量测定人血清或血浆中的总胆汁酸(TBA)的含量。
1.1 包装规格
包装规格见表1。
表1 包装规格
1.2 主要组成成分
主要组成成分见表2。
表2 主要组成成分
注:不同批号的校准品赋值有差异,具体赋值详见靶值单。
2.1 外观
试剂1为黄色澄清液体,目测不得有任何沉淀及絮状悬浮物;
校准品为无色澄清液体,目测不得有任何沉淀及絮状悬浮物;
试剂盒标签标识清晰,外包装完整无损。
2.2 净含量
试剂的净含量应不少于标称量。
2.3 试剂空白
2.3.1 试剂空白吸光度
A405nm下测定空白吸光度应≤0.8000。
2.3.2 试剂空白吸光度变化率
A405nm下测定的空白吸光度变化率(ΔA/min)应≤ 0.0400。
2.4 准确度
与已上市的产品进行比对试验:在[5,180]μmol/L区间内,相关系数r ≥0.975,在[5,40]μmol/L区间内测定的绝对偏差应不超过±6μmol/L,在(40,180]μmol/L区间内测定的相对偏差应不超过±15%。
2.5 分析灵敏度
样本浓度为40μmol/L时,其吸光度变化在0.0200~0.0800之间。
2.6 线性区间
在[5,180]μmol/L区间内,线性相关系数r≥0.99,在[5,20]μmol/L区间内测定的绝对偏差应不超过±2μmol/L,在(20,180]μmol/L区间内测定的相对偏差应不超过±10%。
2.7 测量精密度
2.7.1 重复性
对高、低不同浓度的血清样本或质控品重复测定10次,其测定值的变异系数(CV%)应不大于5%。
2.7.2 批间差
随机抽取三批试剂盒的批间相对极差(R)应不大于10%。
2.8 稳定性
试剂盒在2℃~8℃密封避光保存,有效期为18个月。
在试剂盒有效期满后一个月以内,应符合2.1、2.3、2.4、2.5、2.6、2.7.1的要求。
2.9校准品溯源性
按照《GB/T 21415-2008体外诊断医疗器械生物样品中量的测量校准品和控制物质赋值的计量学溯源性》的要求,提供所用产品校准的来源、赋值过程
以及测量不确定度,试剂盒校准品溯源至企业工作校准品,与积水医疗株式会社校准品比对赋值。