基因表达系列分析(Serial Analysis of Gene Expression,SAGE)技术
- 格式:doc
- 大小:263.00 KB
- 文档页数:7

3基金项目:广西自然科学基金资助项目(No 1桂科回0448016)广西医科大学实验中心外科实验室△通信作者收稿日期22结肠腺癌与正常黏膜组织R P 基因mRNA 表达差异分析3张小龙 高 枫△ 李 卫1(广西医科大学第一附属医院结直肠肛门外科 南宁 530021)摘要 目的:探讨结肠腺癌与正常黏膜组织中核糖体蛋白(Ribosomal p rotein ,RP )基因mRNA 的表达差异。
方法:用基因表达系列分析(Serial analysis of ge ne expre ssion ,SA GE )方法对结肠腺癌组织及正常黏膜组织中核糖体蛋白基因mRNA 的表达进行分析。
结果:在结肠腺癌和正常黏膜组织中共出现了101种RP 基因或RP 类似基因的表达,其中有6种仅在正常黏膜中表达,有17种仅在腺癌中表达,41种R P 基因在癌组织中表达明显升高。
结论:在结肠腺癌和正常黏膜中均有多种R P 基因表达,但两者之间存在明显差别。
关键词 核糖体蛋白基因;表达;结肠腺癌;结肠正常黏膜中图分类号:R73513+5 文献标志码:A 文章编号:10052930X (2008)0320335203THE EXP RESSIO N OF RIBOSOMAL PROTEIN GENES IN ADENOCARCINOMA AND NO R 2MAL M UCOSA OF COLO NZhang X iaolong ,Gao Feng ,Li Wei.(Depa rt ment of Colorectal and A nal Sur gery ,t he Fir st Affiliat ed Hos 2pit al of Guangxi Me dical Univer si t y ,Na nni ng 530021China)Abstract Object ive :To explore t he different expression of ribosomal prot ei n genes in adenocarci noma and nor mal mucosa of colon.Met hods :Two SA GE libra ries of a de noca rcinoma and normal mucosa were genera 2t ed.From t hem ,t he expression of ri bo somal protein gene s was gaine d a nd compared.R esult :The expres 2sion of all t he 101species of ribosomal protein genes or t hose similar to ri bosomal p rot ei n gene s were det ec 2t ed i n adenocarcinoma or normal m ucosa.Among t hem ,17species were det ecte d onl y in a denocarcinoma and 6species only i n normal mocosa.41species ha d si gnificantl y hi gher expression i n cancer.The expres 2sion of t he ot her genes was i n low level in bot h ti ssues or had no difference bet ween t hem.Conclusion :The expression of ri bosomal protein genes was different bet ween colon adenocarci noma and colon normal moco 2sa.The mo st predomi nant diff erence was t hat ma ny RP ge nes ha d significant ly higher expression in cancer 2ous ti ssues.Al so ,6species of RP genes had expression i n normal tissue but not i n cancerous t issue.K ey w or ds ri bosomal p rotein ge nes ;expression ;colon adenocarci noma ;colon normal mocosa 核糖体蛋白是核糖体的组成部分之一,在蛋白质的合成过程中起着极为重要的作用。

基因表达谱分析技术1、微阵列技术(microarray)这是近年来发展起来的可用于大规模快速检测基因差别表达、基因组表达谱、DNA序列多态性、致病基因或疾病相关基因的一项新的基因功能研究技术。
其原理基本是利用光导化学合成、照相平板印刷以及固相表面化学合成等技术,在固相表面合成成千上万个寡核苷酸“探针”(cDNA、ESTs或基因特异的寡核苷酸),并与放射性同位素或荧光物标记的来自不同细胞、组织或整个器官的DNA或mRNA反转录生成的第一链cDNA进行杂交,然后用特殊的检测系统对每个杂交点进行定量分析。
其优点是可以同时对大量基因,甚至整个基因组的基因表达进行对比分析。
包括cDNA芯片(cDNA microarray)和DNA 芯片(DNA chips)。
cDNA芯片使用的载体可以是尼龙膜,也可以是玻片。
当使用尼龙膜时,目前的技术水平可以将20000份材料点在一张12cm×18cm的膜上。
尼龙膜上所点的一般是编好顺序的变性了的双链cDNA片段。
要得到基因表达情况的数据,只需要将未知的样品与其杂交即可。
杂交的结果表示这一样品中基因的表达模式,而比较两份不同样品的杂交结果就可以得到在不同样品中表达模式存在差异的基因。
杂交使用的探针一般为mRNA的反转录产物,标记探针使用32PdATP。
如果使用玻片为载体,点阵的密度要高于尼龙膜。
杂交时使用两种不同颜色的荧光标记不同的两份样品,然后将两份样品混合起来与一张芯片杂交。
洗去未杂交的探针以后,能够结合标记cDNA的点受到激发后会发出荧光。
通过扫描装置可以检测各个点发出荧光的强度。
对每一个点而言,所发出的两种不同荧光的强度的比值,就代表它在不同样品中的丰度。
一般来讲,显示出来的图像中,黄色的点表示在不同的样品中丰度的差异不大,红色和绿色的点代表在不同样品中其丰度各不相同。
使用尼龙膜为载体制作cDNA芯片进行研究的费用要比玻片低,因为尼龙膜可以重复杂交。
检测两种不同的组织或相同组织在不同条件下基因表达的差异,只需要使用少量的尼龙膜。


Serial Analysis of Gene Expression(taken from /SAGE/)Serial analysis of gene expression,or SAGE,is a technique designed to take advantage of high-throughput sequencing technology to obtain a quantitative profile of cellular gene expression.Essen-tially,the SAGE technique measures not the expression level of a gene,but quantifies a”tag”which represents the transcription product of a gene.A tag,for the purposes of SAGE,is a nucleotide se-quence of a defined length,directly3’-adjacent to the3’-most restriction site for a particular restriction enzyme.As originally described[1],the length of the tag was nine bases,and the restriction enzyme NlaIII.Current SAGE protocols produce a ten to eleven base tag[2],and,although NlaIII remains the most widely used restriction enzyme,enzyme substitutions are possible.The data product of the SAGE technique is a list of tags,with their corresponding count values,and thus is a digital representation of cellular gene expression.However,to say that SAGE produces a digital output,is not to imply that no loss offidelity occurs from the conversion of an actual transcript and its expression level to a tag and its count value. Accuracy in both the assignment of tags to genes as well as the ability to quantify a gene’s expression level are sacrificed in order to increase throughput,and therefore increase the speed and lower the cost of analysis.A ten base tag is by no means a perfect representation of a gene’s entire transcript.There will be instances in which two or more genes share the same tag(i.e.,the tag to gene assignment is ambiguous),and instances in which one gene has more than one tag(i.e.,through alternate termination in an individual,and polymorphism in a population,the gene to tag assignment is not specific).And, as if this inherent difficulty in making specific and unambiguous tag to gene assignments wasn’t enough, an entirely acceptable sequencing error rate from the point of view of most sequencing tasks can have several disturbing effects on SAGE tag data,when dealing with such short sequences.So there are,really,two problems to be tackled when dealing with SAGE data in the form of tags and counts.Thefirst deals with insuring that the tags and their counts are a valid representation of transcripts and their levels of expression,and the second,with making valid tag to gene assignments.In consideration of thefirst problem–the valid data problem–sequencing error has the greatest effect.Assuming that there is an average1%per base sequencing error rate,for ten bases,the chance of one or more errors occurring is roughly10%[3].The error,if it occurs,will,of course,lower the correct tag count by one,but will also either increase the tag count of an already established tag by one,or will establish and count a tag which does not,in reality,exist.The former effect is not of great concern when drawing conclusions from tags with relatively high counts,since raising or lowering a tag count by one or two should,overall,have no great effect.The former and latter effects,on the other hand,do much to increase suspicion of the tags with low counts,particularly those with a count of1.Currently, the only way this suspicion has been dealt with has been to remove from the data tags counted only once[4].This may not be an optimal approach,and investigations are currently underway to discover if a better approach might exist[5].In consideration of the second problem–making valid tag to gene assignments–unspecific and ambiguous tag to gene assignments,as well as sequencing error,both play a role in creating confusion. In making tag to gene assignments,a certain degree of messiness is encountered.It would be preferable if specific and unambiguous gene assignments could be made for every experimental derived tag,but this is definitely not the case.The difficulties are several,and begin with the set of sequences from which tags are derived.The transcriptome of Homo sapiens has yet to be entirely sequenced,let alone characterized.Until it is,there is only an incomplete set of sequences from which to derive tags.Next, considering the nature of the roughly1.5million transcript-source sequences stored in GenBank,only about18,000are well-characterized cDNA/mRNA sequences,while the vast majority are expressed sequence tag(EST)sequences.The problem with using EST sequences for the derivation of10base tags is that they are usually only single-pass sequenced,and therefore,have,roughly,an average1%per base error rate.Following the reasoning in the paragraph above,this means a10%chance that a10 base tag will include one or more errors.Considering that tag to gene assignments are based upon the sequence from these tags,it stands to reason that roughly10%of the assignments that are extracted from these sequences will be incorrect.This compounds the”naturally”unspecific and ambiguous tag to gene assignments which are already expected without considering sequencing error.The naturally unspecific and ambiguous tag to gene assignments can be measured by extracting SAGE tags from the17,000or so,nearly error-less,well-characterized cDNA/mRNA sequences in GenBank,and matching those tags to some set of defined gene-units.As noted above,since the transcriptome of Homo sapiens have not yet been sequenced or characterized,an artificial method for defining gene-units must be chosen.This may be as simple as taking the title of the GenBank sequence entry,or as complicated as using a set of gene contigs,or using gene-based sequence clusters,such as the UniGene gene set[6].Tags extracted from these nearly error-less sequences give an idea of baseline specificity and ambiguity before sequences with higher sequencing error rates are considered.A heuristic approach has been used to select only3’EST sequences(using sequence submission label, and polyadenylation signal and tail)before extracting tags and making tag to gene ing the estimation of the percentage of tags due to error(i.e.,10%),a”correction”of these EST-based tag to gene assignments can be made by removing10%of the most rarely occurring tags for a particular gene as well as10%of the most rarely occurring genes for a particular tag.This correction has been performed on a rank-ordering of tag-gene pairings[7],and gene-tag pairings[8].Once this correction is made,combining the tag to gene assignments of the well-characterized sequences with those of the corrected EST sequences gives us as much reliable information as possible,while greatly reducing the effect of erroneous tag to gene,and gene to tag,assignments[9].However,an obvious drawback to this approach is that there is no method to tell the difference between tag to gene assignments due to sequencing error,and rare,naturally occurring assignments,which could occur through rare alternate splicing events or polymorphisms,and these would both be removed.Two tag to gene assignment lists,a.k.a.tag-gene mappings,based upon all human transcript-source sequences in GenBank and the UniGene gene clustering algorithm,have been constructed and are publicly available from this website,in both downloadable and interactive versions.These assignments consist of a reliable mapping and a full mapping.The reliable mapping includes the correction for EST sequencing error;the full mapping does not.In addition,this website provides public access to NCI’s Cancer Genetic Anatomy Project(CGAP)SAGE data from human colon and brain tissues,and a statistical test for differential analysis between data sets(or libraries).Gene expression technologies allow us to confront large amounts of data from which we endeavor to glimpse the inner workings of the cell(transcriptome analyses),or differentiate one type of cell from another(differential expression analyses).Obtaining this data is a difficult task unto itself,but,however it is done,collection of the data is only half the rmation must be forcefully extracted from the data–the cacophony quieted–through assumption,knowledge,intuition and fortune.As is true for all analyses,the pump must be primed before information canflow.Notes and References:1.Velculescu VE,Zhang L,Vogelstein B,Kinzler KW.Serial analysis of gene expression.Science.1995Oct20;270(5235):484-7.2.Zhang L,Zhou W,Velculescu VE,Kern SE,Hruban RH,Hamilton SR,Vogelstein B,Kinzler KW.Gene expression profiles in normal and cancer cells.Science.1997May23;276(5316):1268-72.3.1.0−(0.99)10∼0.104.This empirical approach has been used in SAGE tag-count sets in which roughly250,000totaltags have been sequenced.For SAGE data sets,or combinations of sets with,for example,overone million total tags sequenced,it might be necessary to exclude tags with counts of less than2, 3or more.5.A possible approach is to calculate,and make use of,the expected number(or percentage)ofnearest neighbors(i.e.,one base substitution,insertion or deletion)with a count of one,two,etc., given the number of total tags sequenced,and the actual tags in the data set.6.For more information and data about the UniGene project,see /UniGene.7.The tag to gene rank-ordering is a calculated value consisting of the number of sequences containinga particular tag falling into a particular gene group divided by the total number of sequencescontaining that tag.8.The gene to tag rank-ordering is a calculated value consisting of the number of sequences fallinginto a particular gene group containing a particular tag divided by the total number of sequences falling into that gene group.。

基因表达系列分析技术在植物基因表达分析中的应用朱 莉1,2,常汝镇1,邱丽娟1,2(1中国农业科学院作物品种资源研究所/农业部作物种质资源与生物技术重点开放实验室,北京 100081;2新疆农业大学,乌鲁木齐 830052)摘要:基因表达系列分析技术(S AGE )是一种以测序为基础,采用数字化分析手段,在转录物水平上研究细胞或组织基因表达模式的有效工具。
该技术不仅能够全面地分析特定组织或细胞表达的基因,获得这些基因表达丰度的数量信息,还可比较不同组织、不同时空条件下基因表达的差异,从而发现新基因。
S AGE 技术在植物方面的应用相对较少,但进展很快。
本文着重介绍了S AGE 在植物基因表达分析中的应用,分析S AGE 所存在的问题、改进方法及发展前景。
关键词:S AGE;全基因组表达频谱;差异表达;转录组;功能基因组Application of Serial Analysis of Gene Expre ssion in the Analysis of Gene Expre ssion in PlantZH U Li 1,2,CH ANG Ru 2zhen 1,QI U Li 2juan 1,2(1K ey Laboratory o f Crop G ermplasm &Biotechnology ,Ministry o f Agriculture/Institute o f Crop G ermplasm Resources ,Chinese Academy o f Agricultural Sciences ,Beijing 100081;2Xinjiang Agicultural Univer sity ,Urumqi 830052)Abstract :Serial analysis of gene expression (S AGE )is a powerful sequence 2based tool ,which allows the analysis of gene expression patterns in cell or tissue at the level of transcripts with digital analysis.It can be used not only to over al 2ly analyze the genes that expressed in specific cells or tissues ,in order to get the quantity information of the expressed genes ,but als o to simultaneously com pare the differential genes expressed in different tissues and conditions for gene min 2ing.The application of S AGE is fairly little in plant ,but increases rapidly.In the future S AGE will be m ore and m ore used in plant functional genomies.In this review ,the application and im provement of S AGE in plant genomic research are been introduced.K ey words :S AGE ;Whole 2genome expression profile ;Differential expression ;T ranscriptome ;Functional genome收稿日期:2003202208作者简介:朱 莉(19702),女,新疆乌鲁木齐人,农艺师,硕士研究生,主要从事大豆种质资源评价与利用研究。

第二篇细胞的遗传物质第三章基因表达的分析技术生物性状的表现均是通过基因表达调控实现的。
对基因结构与基因表达调控进行研究,是揭示生命本质的必经之路。
在基因组研究的过程中,逐步建立起一系列行之有效的技术。
针对不同的研究内容,可建立不同的研究路线。
第一节 PCR技术聚合酶链反应(polymerase chain reaction,PCR)技术是一种体外核酸扩增技术,具有特异、敏感、产率高、快速、简便等突出优点。
PCR技术日斟完善,成为分子生物学和分子遗传学研究的最重要的技术。
应用PCR技术可以使特定的基因或DNA片段在很短的时间内体外扩增数十万至百万倍。
扩增的片段可以直接通过电泳观察,并作进一步的分析。
一、实验原理PCR是根据DNA变性复性的原理,通过特异性引物,完成特异片段扩增。
第一,按照欲检测的DNA的5'和3'端的碱基顺序各合成一段长约18~24个碱基的寡核苷酸序列作为引物(primer)。
引物设计需要根据以下原则:①引物的长度保持在18~24bp之间,引物过短将影响产物的特异性,而引物过长将影响产物的合成效率;②GC含量应保持在45~60%之间;③5'和3'端的引物间不能形成互补。
第二,将待检测的DNA变性后,加入四种单核苷酸(dNTP)、引物和耐热DNA聚合酶以及缓冲液。
通过95℃变性,在进入较低的温度使引物与待扩增的DNA链复性结合,然后在聚合酶的作用下,体系中的脱氧核苷酸与模板DNA 链互补配对,不断延伸合成新互补链,最终使一条DNA双链合成为两条双链。
通过变性(92~95℃)→复性(40~60℃)→引物延伸(65~72℃)的顺序循环20至40个周期,就可以得到大量的DNA片段。
理论上循环20周期可使DNA扩增100余万倍。
二、PCR技术的分类及其应用范围1.逆转录PCR 逆转录PCR(RT-PCR)是将RNA反转录和PCR结合而建立起来的一种PCR技术。
其包括两个过程:通过逆转录合成cDNA和对之进行PCR扩增。

一、目的基因克隆的策略有哪些?其理论依据什么?如何根据具体条件,如目的性状的特点,已知控制目的性状的基因的信息合理选择基因克隆的方法?令狐文艳1、主要有以下几个克隆的策略:(1)PCR法分离目的基因:从蛋白质的一级序列分析得到核酸序列的相关信息,设计简并引物,通过对mRNA进行反转录得到cDNA,以cDNA为模板,然后将目的基因通过PCR方法扩增,或者直接从基因组DNA扩增的方法。
(2)核酸杂交的方法:通过对蛋白质的氨基酸序列分析,设计简并引物,通过核酸杂交的方法从基因文库中筛选得到目的基因。
(3)免疫学筛选法分离目的基因:利用免疫学原理,通过目的蛋白的特异抗体与目的蛋白的专一结合,从表达文库中分离目的蛋白基因。
2、若控制该性状的目的蛋白质不容易分离纯化,这PCR方法比较适宜,若蛋白质分离纯化容易,且有现成的基因文库,则后两种方法较为简单。
二、蛋白组学方法克隆目的基因的理论依据是什么?有哪些技术环节?要用到哪些技术?1、理论依据:以分离纯化的目的蛋白为研究起点,通过对目的蛋白的一级结构分析,获得起码的氨基酸序列信息后,反推可能的DNA序列,然后设计引物,从cDNA中将目的基因扩增出来,或者设计核酸探针,通过杂交技术将目的基因从基因文库中筛选出来。
或通过抗体抗原免疫反应从表达文库中将该基因分离出来。
2、技术环节是确定并制备出高纯度的蛋白质。
3、所需要的实验技术有:蛋白质的双向电泳技术,由第一向的等电聚焦电泳和第二向的SDS-PAGE电泳组成;蛋白质氨基酸序列分析。
三、基因组学方法克隆基因的策略有哪些?各有什么特点?如何选择恰当的基因组学方法克隆目的基因?1、基因文库筛选方法通过对基因文库的筛选将目的基因分离出来,一般有两种方法:核酸杂交法,原理是分子杂交; PCR筛选法,通过PCR方法将目的基因分离出来,对于以混合形式保存的文库,先将文库分成几份,每份为一个“反应池”进行PCR反应,待选出阳性池后,将阳性池的混合克隆稀释,然后等量分置 96孔板中,进行横向池及纵向池的PCR反应,然后将阳性菌落群进行稀释,重复上述工作,直到筛出阳性单克隆。

基因表达系列分析技术的原理和流程英文回答:Gene Expression Profiling Technologies.Gene expression profiling technologies are used to measure the expression of thousands of genes simultaneously, providing a comprehensive overview of gene activity in a given sample. These technologies have revolutionized the study of biology and disease, allowing researchers toidentify genes and pathways involved in various biological processes and to diagnose and treat diseases.The two main types of gene expression profiling technologies are:Microarray technology uses DNA oligonucleotides fixedto a solid surface to measure the expression of a large number of genes. mRNA from a sample is labeled and hybridized to the oligonucleotides, and the amount ofhybridization is measured. The intensity of the signal for each gene is proportional to the expression level of that gene.RNA sequencing (RNA-Seq) technology uses high-throughput sequencing to measure the expression of all transcripts in a sample. mRNA from a sample is converted to cDNA and then sequenced. The abundance of each transcript is proportional to the expression level of that gene.Gene expression profiling technologies have a wide range of applications in research and medicine, including:Identifying genes and pathways involved in biological processes.Diagnosing and treating diseases.Developing new drugs and therapies.Monitoring the response to treatment.The general workflow for gene expression profiling experiments is as follows:1. Sample preparation.2. RNA isolation.3. Labeling and hybridization (microarray) or cDNA synthesis and sequencing (RNA-Seq)。

SSR标记技术和ISSR 标记技术雷世勇 2.1 SSR 标记技术。
在真核生物基因组中存在许多非编码的重复序列,如重复单位长度在15~65 个核苷酸的小卫星DNA ,重复单位长度在2~6 个核苷酸的微卫星DNA。
小卫星和微卫星DNA 分布于整个基因组的不同位点。
由于重复单位的大小和序列不同以及拷贝数不同,从而构成丰富的长度多态性。
定义:SSR (全称为简单序列长度多态性标记)也称微卫星DNA ,是一类由几个多为1~5 个碱基组成的基序串联重复而成的DNA 序列,其中最常见的是双核苷酸重复,即 CA n和 TG n ,每个微卫星DNA 的核心序列结构相同,重复单位数目10~60 个,其高度多态性主要来源于串联数目的不同。
根据微卫星重复序列两端的特定短序列设计引物,通过PCR 反应扩增微卫星片段。
由于核心序列重复数目不同,因而扩增出不同长度的PCR 产物,这是检测DNA 多态性的一种有效方法。
微卫星序列在群体中通常具有很高的多态性,而且一般为共显性,因此是一类很好的分子标记。
SSR分子标记的应用举例――鹅掌楸种属及杂种的分子标记北美鹅掌楸EST序列中开发的EST-SSR引物引物筛选物种特异性扩增引物特异性验证 SSR 标记技术的特点有:(1)数量丰富,广泛分布于整个基因组;(2)具有较多的等位性变异;(3)共显性标记,可鉴别出杂合子和纯合子;(4)实验重复性好,结果可靠;(5)由于创建新的标记时需知道重复序列两端的序列信息,因此其开发有一定困难,费用也较高。
SSR 标记的应用目前已利用微卫星标记构建了人类、小鼠、大鼠、水稻、小麦、玉米等物种的染色体遗传图谱。
这些微卫星标记已被广泛应用于基因定位及克隆、疾病诊断、亲缘分析或品种鉴定、农作物育种、进化研究等领域。
2.2 ISSR 标记技术 ISSR 即(内部简单重复序列),是一种新兴的分子标记技术。
它是建立在1994年发展的一种微卫星基础上的分子标记。

基因组学与应用生物学,2009年,第28卷,第6期,第1204-1210页Genomics and Applied Biology,2009,Vol.28,No.6,1204-1210专题介绍Review基因表达系列分析法(SAGE)的改进及其在植物功能基因组研究中的应用张振乾谭太龙肖钢官春云﹡湖南农业大学农学院,国家油料改良中心湖南分中心,长沙,410128*通讯作者,guancy2000@摘要基因表达系列分析方法(SAGE)是一种新的基因表达分析方法,与基因芯片技术一样具有高通量的特点,可测定特定组织的基因表达水平,在全基因组水平上同时定量检测数万个基因表达模式;可在未知目的基因的前提下,分析来自一个细胞的全部转录本信息;对已知或未知基因表达进行定性和定量分析。
目前,虽然在疾病、发育、细胞凋亡、药物筛选等多个领域已有利用SAGE 方法进行的研究,但该方法在植物功能基因组研究中的应用相对较少。
本文主要综述了该方法在RNA 用量、PCR 循环次数、SAGE 效能和可靠性、标签长度和未知标签分析等方面的改进及其在植物中构建SAGE 文库、筛选新基因、基因表达图谱分析等方面的应用,从而为其在植物功能基因组研究中的进一步应用提供理论参考。
关键词基因系列分析方法(SAGE),功能基因组,基因表达图谱分析The Modification of Serial Analysis of Gene Expression (SAGE)and its Ap-plication in Plant Functional Genome ResearchZhang ZhenqianTan Tailong Xiao Gang Guan Chunyun ﹡Hunan Agricultural University,Hunan Branch of National Oilseed Crops Improvement Centre,Changsha,410128*Corresponding author,guancy2000@ DOI:10.3969/gab.028.001204Abstract Serial analysis of gene expossion (SAGE)is a kind of innovative gene expossion analysis method ,which possesses the same characteristic of high throughput as Genechip.This techniques can determine the level of gene expression in some special tissues and can quantitatively detect expression patterns of thousands of genes simultaneously on the whole genome level.And it also can be used to analyze all transcripts information from a cell without knowing target gene.Furthermore,This analytical method can carry out the qualitative and quantita-tive analysis for the expression of known or unknown genes.At present,SAGE has been widely used in many fields,such as disease,development,apoptosis and drug screening etc.,whereas it is relatively less been applied in plant functional genome research.In this paper,we mainly reviewed the modification of this approach on RNA dosage,PCR cycles,the efficacy and reliability of SAGE,the label length and unknown label analysis and so on.At the same time,we also summarized its application in plant functional genome research,for instance,the con-struction of SAGE library,screening of new genes and gene expression profiling analysis,which would provide theory reference for its further application in plant functional genome research.Keywords Serial analysis of gene expression (SAGE),Functional genome,Gene expression profiling analysis /doi/10.3969/gab.028.001204基金项目:本研究由国家973计划(2006CB101600)资助利基因表达系列分析方法(serial analysis of gene expression,SAGE)是基于poly A +RNAs 剪切得到的短标签和标签末端间的两两连接组成的串联体分析方法(Richards et al.,2004)。
SAGE 技术MRNA 结合到微珠子上(Microscopic Bead and mRNA)mRNA 转录成DNA(mRNA binds to bait and is copied into DNA)用酶切开DNA的一小段(An enzyme cuts the DNA)另一个酶定在DNA末端以便切下一小段(An enzyme locks onto the DNA and cuts off a short tag),这一小段就被视为这个基因的标签两个标签连在一起(Two tags are linked together)在末端的定位分子被切掉(Enzymes cut off the "Docking Molecules")都连成一条线(Di-Tags are combined into large concatemers)DNA上所携带的遗传信息,需要通过RNA为中介体,合成出组织和正常生理功能所需要的蛋白质,这个过程被称为基因的表达。
在生物体中不同的组织和器官所表达的基因群是不一样的,我们把基因群的表达状况称为基因表达谱。
目前,高通量地研究基因表达谱的方法主要有两种,即生物芯片和基因表达串联分析(serial analysis of gene expression, SAGE)。
基因芯片所能检测的基因必须是已知的基因,放在芯片上几种基因的探针就只能检测这几种基因的表达谱;相比之下,SAGE能以远高于DNA芯片的精确度和重复性来检测在病理条件下基因表达谱的改变,而不必考虑所检测的基因是已知的还是未知的。
因此在检测疾病相关的新基因,特别是无法用基因芯片进行检测的低表达量致病基因时,SAGE是目前的最佳手段,无可取代。
SAGE技术为Genzyme公司所拥有的专利技术。
其技术简介如下:SAGE技术得以建立的理论基础首先,一段来自于任一转录本特定区域的"标签"(Tag),即长度仅9-14bp的短核苷酸序列,就已包含足够的信息以特异性地确定该转录本。
第8章基因表达数据分析基因芯片或DNA微阵列等高通量检测技术的发展,可以从全基因组水平定量或定性检测基因转录产物mRNA,获取基因表达的信息。
由于生物体中的细胞种类繁多,同时基因表达具有时空特异性,因此,基因表达数据要比基因组数据更为复杂、数据量更大、数据的增长速度更快。
基因表达数据中蕴含着基因调控的规律,可以反映细胞当前的生理状态,例如(??)是否恶化、(??)是否对药物有效等。
对基因表达数据的分析是生物信息学的重大挑战之一,也是DNA微阵列能够推广应用的关键环节之一。
基因表达数据分析的对象是在不同条件下,全部或部分基因的表达数据所构成的数据矩阵。
通过对数据矩阵的分析,回答一些生物学问题,例如,基因的功能是什么?在不同条件或不同细胞类型中,哪些基因的表达存在差异?在特定的条件下,哪些基因的表达发生了显著改变,这些基因受到哪些基因的调节,或者调控哪些其它的基因?哪些基因的表达是条件特异性的,根据它们的行为可以判断细胞的状态(正常或癌变)????等等。
对这些问题的回答,结合其他生物学知识和数据有助于阐明基因的调控路径和基因之间的调控网络。
揭示基因调控路径和网络是生物学和生物信息学共同关注的目标,是系统生物学(Systems Biology,在附录中增加解释条目!)研究的核心内容。
目前,对基因表达数据的分析主要是在三个逐渐复杂的层次上进行:1、分析单个基因的表达水平,根据在不同实验条件下,该基因表达水平的变化,来判断它的功能,例如可以确定肿瘤类型特异基因。
采用的分析方法可以是统计学中的假设检验等。
2、考虑基因组合,将基因分组,研究基因的共同功能、相互作用以及协同调控等。
多采用聚类分析等方法。
3、尝试推断潜在的基因调控网络,从机理上解释观察到的基因表达谱。
多采用反工程的方法。
本章首先介绍基因表达数据的来源和预处理方法;然后介绍基因表达数据分析的主要方法,即表达差异分析和聚类分析;最后简单介绍从基因表达数据出发研究基因调控网络的一些经典模型。
基因表达系列分析技术的原理及应用进展262国外医学分子生物学分册2000年第船卷第5期一表达系列分析技术的原理及应用进展综述粟采萍审阅第三军医大学全军复合伤研究所(重庆,400038)摘要生物体的特性由内在基因表达决定个体发育和疾病发生与基因选择性表达密切相关,因此,要了解生命活动和疾病发生,发展的分子机制,就必须椿人了解基因表达的时空性和差异性基因表达系列分析技术(SAGE)是新近建立的能够定性和定量研究基因表达的有效工具,不仅可提供基因组表达丰度教完整的数量信息,而且还可帮助寻找和发现新基因, 关键调苎里懿差型坌堑7sAGr舀谪f镗目前,用于研究基因表达的方法较多,主要有cDNA消减杂交(subtractionhybridiza—tion,SH)'1],mRNA差异显示(differentialdisplay,DD),代表性差异分析法(repre—sentationaldifferenceanalysis,RDA)[,EST(expressedsequencetag)序列鉴定],cDNA微阵列杂交技术(cDNAmicroarray)和基因表达序列分析(serialanalysisofgeneexpression,SAGE)_6技术及上述几种技术的组合基因鉴别组合程序" (integratedprocedureforgeneidentifiea—tion,IPGI)口].以上技术都是分析差异基因表达的有效技术,但各有优缺点.EST与DD都可用来寻找新基因,但前者耗资巨大,后者又有难于克服的假阳性且只能发现有限的基因;微点阵杂交能同时分析大量样本,只能分析已知基因;IPGI叉过于繁琐.而SAGE技术能较完整地获得基因组表达丰度的数量信息,适合于比较不同细胞株和不同发育状态以及不同疾病情况下的基因表达情况.同时还可发现新基因.该技术中锚定酶(anchor ingenzyme)和fS型标签酶(tagglngen—zyme)的选择具有多样性,使得该策略有很大的灵活性.因此,任何一个具备PCR与DNA自动测序条件的实验室都可以开展此技术.本文就该技术的原理和应用进展作简要介绍.1基因表达系列分析的基本原理和实验流程1.1SAGE的原理SAGE技术基于两个依据:①一个来自转录物内特定位点的9~1Obp短核苷酸序列,包含了鉴定一个转录物足够的信息.9bp 核苷酸序列可以区别=262144转录物,而人类基因组编码的转录物仅有80000个, 所以理论上每一个9碱基标签能够代表一种转录物的特征序列.②将9bp的标签集中在一个克隆中进行测序,得到大量的短核苷酸序列以连续的数据输入计算机中,通过相应的软件和联网分析,就能对数以万计的mR NA转录物进行分析.1.2SAGE的基本实验流程如下①提取RNA,分离mRNA,以生物素化寡核苷酸(dT)为引物反转录合成cDNA,用限制性内切酶(锚定酶,anchoringenzyme AE)酶切.~bp识别位点的限制性内切酶,平均可切割4':256碱基,大多数转录物均大于此范围.因此,选用4bp识别位点的限制性内切酶可保证每一种转录物上至少有一个酶切位点.通过链霉抗生物素蛋白磁珠收集mRNA的polyA尾与最近酶切位点之间的片段②将eDNA分成两部分,连接接头A或B.接头含有标签酶(taggingenzyme,TE)的酶切位点.它具有在距识别位点20个碱基位置以外酶切的特点.接头的结构为引物A/ B序列+标签酶识别位点+锚定酶识别位点.用标签酶酶切产生含有接头约9~lObp 的cDNA片段,收集带有linkerl,linker2的酶切片段,用连接酶连接,形成双标签,以引263?物A和B进行PCR扩增③用锚定酶酶切PCR产物,用聚丙酰胺凝胶分离双标记序列,并用连接酶构建成多联体将多联体克隆进滟l序载体进行测序分析.一般每一个克隆最少有lO个标签序列.测序结果用SAGE软件分析,并与Genbank,ESTdatabases等资料库比较分析,得到转录物丰度的数量信息和新的表达基因.(SAGE的方案见图1).…c^TG…弛£liE的c硎^克隆图l基因表达系列分折(SAGE)示意图X扣O分别表示不同标签曲核苷酸顺序2基因表达系列分析技术的应用与展望1995年V elculescu等首先在CSeienee》杂志上发表了用SAGE技术对人胰腺基因表达进行分析的结果.在分析的1000个标签中, 95%以上的标签能代表独有的转录物.转录水平依标签出现频率(共840个)分为4类:①超过3次共380个,占45.2;②出现3次共4j 个,占5.4;③出现两次共64个,占7.6;①仅出现1次351个,占41.8.所以SAGE出lA£_-AE能够直观地展示生物体基因表达的数量信息, 对基因进行量化分析.同时用13bp(9bp+锚定酶的4个bp)分离了4个未确定标签所对应的转录物,以此为探针对胰腺cDNA文库进行筛选,找到了一个以前未知特征的胰腺转录物其结果表明SAGE技术不仅可以对基因表达进行定量和定性分析,还可发现新的基因1997年V eleuleseu等又报道了用SAGE方法对不同生长期酵母转录分析数据用于构建染色体的表达图谱.共分析60633个转录物,4665个基因在每个细胞中的表达水平为0.3~200个拷贝数,其中20184个处于对数生长期}20034个处于S期}20415处于G,/M期.1981个基因功能已明确,而2648个基因未被报道.利用基因的表达信息与基因组图谱融合绘制的染色体表达图谱,使基因表达与物理结构联系起来,有利于基因表达模式的研究.1998年Chen-.等用SAGE对比分析大鼠肥大细胞株(RBL一2H3)在静止期和激活期基因表达的特点.40759个转录物中的11300 个基因被分析,激活的肥大细胞单核细胞趋化蛋白(MCP),TGF—B,M—CSF的基因表达分别高于静止期肥大细胞的20~8倍左右,这与已往的报道结果一致.同时发现擞活的肥大细胞以前未报道的表达基因,如周期素G(Cyclin G),肾上腺索(adrenodoxin),前释放素(pre- prorelaxin),促丝裂原活化蛋白激酶激酶(MAPKK),rVH6,dsPTP等基因的表达,对阐明变态反应性疾病的分子机制和治疗提供理论依据.Kenji[10J等用SAGE系统分析了非小细胞肺癌的转录特点,226000个标记序列被分析,代表了43254个转录物,其中142个转录物在肿瘤细胞过度表达,比正常组织高出约10倍,Northern杂交证实有15个转录物在肿瘤细胞异常丰富,其中PGP9.5,B—myb和人mutt基因在原发性肺癌中表达丰富.而其它组织的原发性肿瘤表达较低,为肺癌的诊断,治疗提供了新的途径.1999年deWaard【]等用SAGE分析动脉粥样硬化刺激物(氧化的低密度脂蛋白)对内皮细胞作用后不同时相点的基因表达情况. 12000标记序列中有5出现了表达的差异.共有56个基因表达不同,42个基因是已知的.如:IL一8,MCP一1,VCAM一1,PAI一1.Gro—a,Gro一口和选择素E(E—selectin),用Northern印迹验证上调表达的基因,从分子水平探讨了动脉粥样硬化形成的机制.1999年1月29日至2月1日在荷兰举行了SAGE研究专题讨论会,提交会议论文34篇",主要是欧美10个国家参加,报道了用sAGE技术研究肿瘤,肝硬化,糖尿病,动脉粥样硬化,先天性甲状腺功能低下和唐氏综合征(Down'Ssyndrome)等的研究结果,均获得不同程度的新发现,并提出一些创新性见解通过互联网等查询从1995年到目前,用SAGE研究正式发表的文献有20余篇,主要发表在《Science》,《Cell》及《NueleieAcids Res》杂志上.由于sAGE可以提供基因表达定量和定性的结果,用不同识别位点的锚定酶与酶切位点距离识别因子5~20bp的IIS酶组合,使这一策略具有很大的灵活性.因此.该方法一经问世,便受学者青睐,相关文献报道逐年递增.近年来,拢国还未见有这方面的正式报道.但有部分学者已开始了这方面的工作.随着人类基因组计划的完成,确定高等生物包括人类在内的基因组序列,已是现实可行的目标.因此,对生物发生,发展及疾病的形成过程中基因表达的变化和差异基因的鉴定以及新基因功能的认识和调控就显得尤为重要.尤其是疾病基因鉴定将为疾病诊断,治疗提供有用的标记.随着SAGE技术的不断完善和发展,将对分子生物学的发展起到巨大的推动作用.参考文献1TravisGHatProcNatlAcadSctUSA.1988;85:16962LiangPat.Science,1992:257:9673LisitsynNetat.Science,1993;259:9464AdamsMeta1.Nalure,1992;355:6325LaskariDeta/.ProcNatlAcadScIUSA,1997;94{13056V elculescuV eta/.Science.1995{270:4847WangSMeta1.ProcNarlAcadSctUSA.1998:95:119098V elcutescuV eta/.Cell,1997:88{2439ChenHXetaLJExpMed,1998;188:I65710KenjiHeta1.CancerRes.1998j58:5690nDeWaaTdV at.Gone,1999}226(1):112TheAbstractionofthemeetingofSAGErk—shop.Hilversum,TheNetherlands1999,1,27(1999-09-23啦蕊) A瑚表…华西医科大学附属第一医院医学遗传学教研室(成都,610041)播蔓甲基化修饰是脊推动物DNA唯一的自然掺饰方式.动物基因组甲基化与基因表达密切相美,DNA甲基化通过与反式作用因子相互作用或通过改变染色质结构而影响表达,在X染色体失活,基因组印记,肿瘤发生发展中起重要作用.共董调DNA甲基化'cpG岛;甲基转移酶;基因表达调控DNA甲基化是哺乳动物DNA最常见的复制后调节方式之一,也是脊椎动物DNA唯一的自然化学修饰方式,它是由甲基转移酶介导,将胞嘧啶(c)变为5一甲基胞嘧啶(5~mC)的一种反应.哺乳动物DNA甲基化发生在CpG二核苷酸的胞嘧啶上,哺乳动物基因组约有5~10是CpG位点,其中7O~8O为mCpG.CpG二核苷酸大部分集中于异染色质区,呈高度甲基化,其余则散布在基因组中,平均每100kb有一个0.5~2.5kh的CpG岛.基因组中约一半基因(主要是看家基因和约40的组织特异基因)的调控序列中存在CpG岛,散在的CpG岛在正常情况下大部分是非甲基化的,而在失活x染色体,印记基因和非表达的组织特异基因中则是甲基化的"J.动物基因组甲基化与基因表达密切相关,即基因在不表达的组织中被甲基化,而在特异表达的组织中非甲基化甲基化类型通过从新甲基化(dellOVOmethylation)和去甲基化(demethylation)形成.在前囊胚期早期,基因组经历了去甲基化,而在种植期经历一个从新甲基化的过程.此时大部分CpG 岛被甲基化在以后的发育阶段,组织特异性基因通过选择性的去甲基化而形成特异的细胞表达类型.1DNA甲基化与基因表达1.1甲基他和去甲基化DNA甲基化发生在很短的时期,其后通过甲基转移酶活性来维持.目前已知且基因已被克隆的甲基转移酶如MTase/Dnmtl显示两种活性:一是在半甲基化的双链上对稚性地保持甲基化(maintence methylation);二是从新甲基化(denov0 methylation).在体外或哺乳动物细胞中,甲基转移酶从新甲基化的作用要比维持甲基化的作用弱.哺乳动物可能存在去甲基化酶.这种酶可能是一种糖基化酶,核酸内切酶或真正的去甲基化酶,Weiss"等的研究显示,在发育过程中去甲基化酶存在于所有细胞,酶中包含有RNA成分,可被一个结合蛋白抑制.现。
【交流】核酸版12月份明星技术:Serial Analysis of GeneSerial analysis of gene expression (SAGE) is a powerful tool that allows the analysis of overall gene expression patterns with digital analysis. Because SAGE does not require a preexisting clone, it can be used to identify and quantitate new genes as wellas known genes.SAGE 技术在理论上来说可以检测到一个细胞内所有转录体的表达,而且可以给每一个转录体定量,不管它是低丰度还是高丰度。
SAGE和基因芯片技术一样,具有高通量、平行性检测细胞内基因表达谱的特点。
但SAGE可在未知任何基因或EST序列的情况下对靶细胞进行研究,这一点是基因芯片技术所不具备的。
目前关于SAGE技术的文章在国外发表很多,但感觉国内搞SAGE研究的不是很多。
本版特地在继10月份推出基因芯片技术大讨论之后,推出12月份明星技术:SAGE(11月由于投票等原因未能推出明星技术讨论,sorry)。
希望大家踊跃参加!加分原则:1. 你可以在此跟帖提问。
如果你的问题有一定深度,引发战友们积极讨论和思考。
加1-2分。
2. 回答问题或介绍经验。
可加1-2分。
3. 精品论文导读(Cell,Nature,Science,Nature Genetics,PNAS等杂志上的更易加分),可加1-3分,即给出全文链接,且深入浅出介绍文章的主要内容加1分;且对文章的实验设计、结果和本文可能开展后续工作的介绍加2分;同时用流线图画出文章的技术路线、思路或结构的加3分。
站友们在发知识性的帖子时,请注意标明“原创”或“转帖(及其来源)”。
基因表达系列分析及其在寄生虫学研究中的应用
李巧丽;张志明;乔中东
【期刊名称】《中国寄生虫学与寄生虫病杂志》
【年(卷),期】2007(25)6
【摘要】基因表达系列分析(serialanalysisofgeneexpression,SAGE)是一种高通量、快速定量分析真核细胞基因表达信息的技术。
它通过抽取距离每个mRNA中3′端polyA尾最近的一个锚定酶识别位点下游的10~14bp片段作为其代表性标签,连成长串,克隆到载体中,进行高效测序,统计出研究对象中的mRNA的种类和丰度。
SAGE技术不仅能够全面地分析特定组织或细胞表达的基因并得到这些基因表达丰度的数量信息,还可比较不同组织、不同时空条件下基因表达的差异。
本文将综述基因表达系列分析技术的原理、特点、方法、进展及其在寄生虫学研究中的应用。
【总页数】6页(P504-509)
【关键词】基因表达系列分析;mRNA表达谱;寄生虫学
【作者】李巧丽;张志明;乔中东
【作者单位】上海交通大学生命科学技术学院
【正文语种】中文
【中图分类】R38
【相关文献】
1.基因表达系列分析的原理、进展及在肿瘤研究中的应用 [J], 梁光萍;苏踊跃;罗向东
2.基因表达系列分析技术在消化道肿瘤研究中的应用 [J], 宋向凤;王辉
3.基因表达系列分析技术在真菌功能基因组学中的应用 [J], 殷朝敏;雷靖行;陈叶叶;马爱民
4.基因差异表达分析技术在寄生虫学中的应用研究进展 [J], 夏艳勋;李国清;苑纯秀;赵传璧;林矫矫
5.基因表达系列分析法(SAGE)的改进及其在植物功能基因组研究中的应用 [J], 张振乾;谭太龙;肖钢;官春云
因版权原因,仅展示原文概要,查看原文内容请购买。
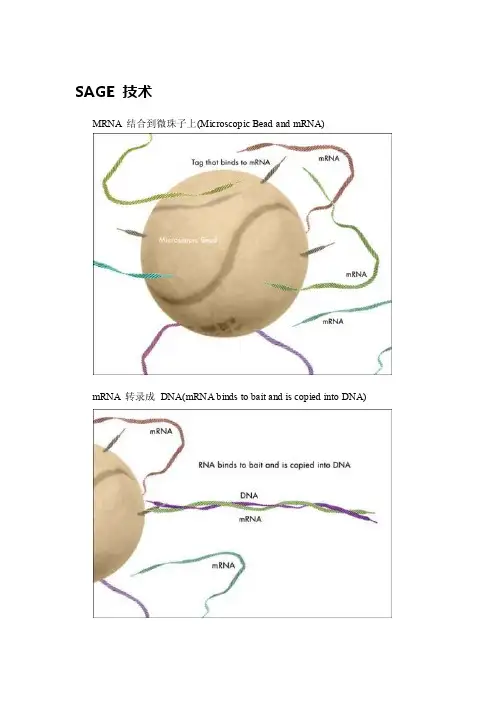
SAGE 技术
MRNA 结合到微珠子上(Microscopic Bead and mRNA)
mRNA 转录成DNA(mRNA binds to bait and is copied into DNA)
用酶切开DNA的一小段(An enzyme cuts the DNA)
另一个酶定在DNA末端以便切下一小段(An enzyme locks onto the DNA and cuts off a short tag),这一小段就被视为这个基因的标签
两个标签连在一起(Two tags are linked together)
在末端的定位分子被切掉(Enzymes cut off the "Docking Molecules")
都连成一条线(Di-Tags are combined into large concatemers)
DNA上所携带的遗传信息,需要通过RNA为中介体,合成出组织和正常生理功能所需要的蛋白质,这个过程被称为基因的表达。
在生物体中不同的组织和器官所表达的基因群是不一样的,我们把基因群的表达状况称为基因表达谱。
目前,高通量地研究基因表达谱的方法主要有两种,即生物芯片和基因表达串联分析(serial analysis of gene expression, SAGE)。
基因芯片所能检测的基因必须是已知的基因,放在芯片上几种基因的探针就只能检测这几种基因的表达谱;相比之下,SAGE能以远高于DNA芯片的精确度和重复性来检测在病理条件下基因表达谱的改变,而不必考虑所检测的基因是已知的还是未知的。
因此在检测疾病相关的新基因,特别是无法用基因芯片进行检测的低表达量致病基因时,SAGE是目前的最佳手段,无可取代。
SAGE技术为Genzyme公司所拥有的专利技术。
其技术简介如下:
SAGE技术得以建立的理论基础
首先,一段来自于任一转录本特定区域的"标签"(Tag),即长度仅9-14bp的短核苷酸序列,就已包含足够的信息以特异性地确定该转录本。
例如:一个9碱基的序列能有49=262144种不同的排列组合,而人类基因组据估计仅编码80000种转录本,因此在理论上每一个9碱基标签就能够代表一种转录本的特征序列。
第二,如果将短片段标签相互连接、集中形成长的DNA分子,则对该克隆进行
测序将得到大量连续的单个标签,并能以连续的数据形式进行处理,这样就可对数以千计的mRNA转录本进行批量分析。
第三,各转录本的表达水平可以用特定标签被测得的次数进行定量。
SAGE的主要实验阶段:
1)以biotin-oligo dT为引物将mRNA反转录合成双链cDNA,以一种锚定酶(Anchoring Enzyme, AE)进行酶切后,收集其cDNA的3'端部分。
锚定酶通常为识别4碱基位点的III类限制性内切酶,如Nla III、Sau3A I等。
由于大多数mRNA 的长度要大于44=256个碱基,因此使用这种锚定酶可以保证在每一个转录本上至少有一个酶切位点。
2)将收集的3'端cDNA等分为两部分,分别同接头A、B相连接。
每一种接头的结构都由PCR扩增引物A或B的序列--标签酶识别位点--锚定酶识别位点三部分组成。
标签酶(Tagging Enzyme, TE)是一种IIS类限制性内切酶,如BsmF I等,它在距识别位点下游约20个碱基的位置切割DNA双链。
3)连接产物以标签酶酶切后用Klenow酶补平5'突出端,得到两组分别带有接头A、B的短cDNA片段(约50个碱基)。
混合并连接两组短cDNA片段,形成一个约100个碱基的双标签体(ditag)群,并以引物A和B对其进PCR扩增。
4)用锚定酶切割扩增产物,分离纯化去除接头后的双标签体(约26个碱基),并使之相互连接成为大片段的连接体,克隆至质粒载体内形成一个SAGE文库以备集中测序。
5)对测序得到的标签数据进行分析处理。
在所测得序列中的每个双标签体之间由锚定酶序列相间隔,一般一个测序反应的结果可得到约20个双标签体,亦即包含了约40个转录本的信息。
由于双标签体的长度基本相同,不会产生PCR扩增的偏态性;同时数量和种类极大的转录本群体使得由同两个标签重复连接成双标签体的可能性极小,因此通过计算机软件的分析统计能够相当精确地得到上千种基因表达产物的标签序列及其丰裕度。
对于在已知的数据库中找不到相同序列的标签,还可以利用约13个碱基的寡核苷酸探针(锚定酶识别位点+标签序列)对cDNA文库进行筛选,以寻找新基因。
SAGE可以检测不同细胞间已知基因表达的具体差异(可精确到每个细胞中大约有多少拷贝),可建立较全面的基因表达谱,系统地分析基因表达的差异。
基因是生物制药事业的源头,生长点和制高点,源于基因的技术拓展将是21世纪制药企业开发新品的基础,基因研究现在成为全球瞩目的焦点。
目前,世界上各大制药,化工和农业公司都在积极地进行改组、合并和建立新联盟,以通过基因相关的研究和开发加强自己的竞争实力。
尽管基因产业所需的投资数目非常大,探索工作也非常艰辛(比如分离囊性纤维病变基因花了十年时间,耗资1.5亿美元以上),但一旦找到一个能够编码重要功能的蛋白的基因后,其回报将是无比丰厚的,发现者获得该基因的专利。
科研人员可以之进行相关研究并设计相关的防治药物,医药公司可在专利期满之前获取市场巨额垄断利润。
可见一个基因可以成为一个企业,甚至带动一个产业。
基因是一种有限资源,人体共有约3万4千个基因,人类基因组只有一套,世界各国投入巨资寻找基因的研究实为一场"基因抢夺战""基因侦探们"在这3万4千个基因中逐个探索,谁占有较多的基因专利,谁就将在人类基因的商业开发方面抢得先机。
SAGE技术研究基因表达谱是1995年才开始的工作,有相当高的技术难度,目前全世界仅有数家实验室能完全掌握SAGE技术。
用SAGE技术得到的数据比DNA 芯片有高得多的可靠性和再现性,利用其可高效率地进行基因表达谱数据库的收集工作。
通过后续的生物信息学处理、转基因等研究方法,可以高效率地找到疾病相关基因,开发出疾病诊断的新方法,为新药研究和新治疗法的开拓提供坚实可靠的基础。
两者比较
基因表达序列分析(SAGE)和大规模平行测序技术(MPSS)是两个基于短标签测序的方法。
它们可以通过短的序列标签(SAGE短的序列标签为10或17bp,MPSS大规模平行测序技术短的序列标签为13或20 bp),能够量化基因的表达,这些标签在多聚腺苷酸转录物内,毗邻NlaIII(SAGE)或DpnII(MPSS)的3’端。
SAGE和MPSS实验的结果是短序列集,某一特定标签的频率代表对应转录物的丰度。
虽然SAGE和MPSS可以产生类似的输出结果,即一系列的标签,但它们的实验方案是完全不同的。
例如,SAGE使用传统的克隆及DNA测序方法,MPSS则基于酶消化和杂交,利用新的克隆和专属的平行测序方案。
因此,SAGE平均输出包含10万个标签,而MPSS能
够输出包含超过1,000,000个标签。
一个在SAGE和MPSS数据分析中的重要步骤就是正确指定/定位标签到基因。
基本上有三种策略来完成这个过程:
(1)基于数据库的注解,数据库通过内部电脑方案构建;
(2) 注解,标签对标签,通过网络站点完成,如SAGE Genie数据库和SAGEmap数据库。
SAGEmap是美国国家癌症研究所(NCI)提供的一个在线SAGE分析工具,其数据库包含多种正常组织、癌前病变组织和肿瘤组织的SAGE文库。
SAGE Genie数据库在SAGEmap 的基础上提供了更为友好的界面,通过查询SAGE Genie数据库,可以获知基因在正常组织和癌变组织中的相对表达量;
(3) 利用参考数据库大规模注解。
SAGE分析的另一个重要步骤是识别每个样本中的不同标签。
为此,有几种方法可以采取,与Baggerly等人、Thygesen和Zwinderman以及Zuyderduyn所描述的一样。
SAGE的显著特点是快速高效地、接近完整地获得基因组的表达信息,它可以定量分析已知基因及未知基因表达情况。
在疾病组织、癌细胞等差异表达谱的研究中,SAGE可以帮助获完整转录组学图谱,发现新的基因及其功能、作用机制和通路等信息。
MPSS是对SAGE的改进,它能在短时间内检测细胞或组织内全部基因的表达情况,是功能基因组研究的有效工具。
但它需要的配套软硬件较为昂贵,目前国内外相关的应用报道不多。
MPSS技术对于致病基因的识别、揭示基因在疾病中的作用、分析药物的药效等都非常有价值,该技术的发展将在基因组功能及其相关领域研究中发挥巨大的作用。
SAGE和MPSS技术自出现以来,就被人们广泛应用(特别是SAGE技术)。
许多公布的实验,尤其是癌症研究实验都已经开始用这些方法来分析基因的整体表达。
最早对人类癌基因组进行全面基因分析的技术是SAGE。
有三个特点令SAGE和MPSS成为癌症中基因表达分析的有效方法
(1)SAGE与MPSS不用事先选择基因进行研究就可以提供mRNA群体的特点,这样可以发现新基因,例如致癌基因;
(2) 在一个实验中获得的数据可以直接比较任何其它实验室的数据,或比较现有公共数据库的数据,允许一个大规模的基因表达比较;
(3) 生成的数据,标签的频率是以数字格式展现,能够稳健地进行基因表达统计分析。