进化树(Phylogenetic_tree)
- 格式:ppt
- 大小:825.50 KB
- 文档页数:33




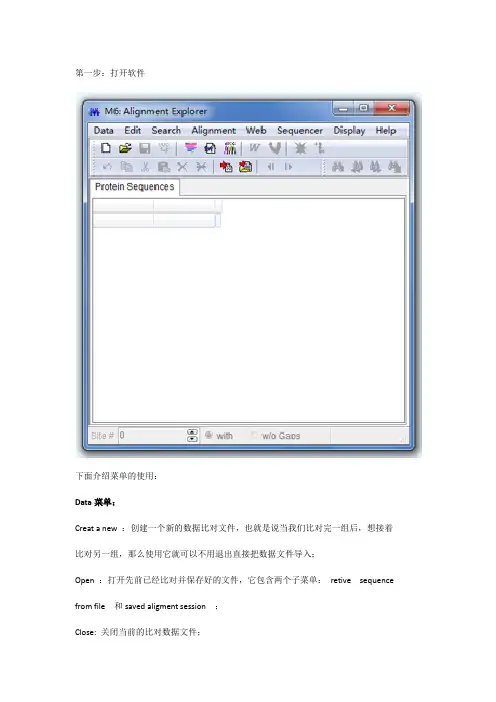
第一步:打开软件下面介绍菜单的使用:Data菜单:Creat a new :创建一个新的数据比对文件,也就是说当我们比对完一组后,想接着比对另一组,那么使用它就可以不用退出直接把数据文件导入;Open :打开先前已经比对并保存好的文件,它包含两个子菜单:retive sequence from file 和saved aligment session ;Close: 关闭当前的比对数据文件;Save session :保存当前比对结果,可以给比对的结果一个文件名;Export alignment :将当前的序列比对结果输出到指定文件,有两种输入格式可供选择:MGTA 和FASTA.DNA sequence :使用它来选择输入的数据DNA 序列,这里需要说明的是如果你输入的数据是氨基酸序列的话,比对窗口只显示一个标签,若是DNA 序列的话则显示两个标签,一个是DNA 序列的,另一个是氨基酸序列的。
Protein sequences :选择输入的氨基酸序列,选择后,所以的位点就被当作氨基酸残基位点来对待。
Translate/untranslate :只有比对的序列是编码蛋白的DNA序列的时候才可用。
它可以根据指定的遗传密码表将DNA 序列翻译成特定的氨基酸序列。
Select genetic code table :使用它将编码蛋白的DNA 翻译成特定的蛋白序列。
R everse complement :将选择的一整行的DNA 序列变为与之互补配对碱基序列。
Exit alignment explorer :退出序列比对的资源管理窗口Edit 菜单:使用这个菜单可以对我们的比对序列进行想要的一些编辑工作具体为Undo:撤销上一步操作;Copy:复制;Cut:剪切;Paste:粘贴;这三个操作都可以只针对一个碱基或氨基酸残基也可以是一段甚至是整个序列;Delete:从比对表格中删除一段序列;Delete gaps:去掉序列中的空缺;Insert blank sequence:重新插入一空行;标签和序列都是空的;Insert sequence from file :从已保存的文件中插入新的序列;Select sites :选择一列序列,与点击比对表上方的灰白空格作用类似;Select sequence:选择一行序列,与点击比对表格左侧的标签名作用类似;Select all:全选;Allow base editing :只读保护,只有选择后才能对序列进行编辑操作,否则所以的序列为只读格式,不能进行任何编辑操作。


极为详细的建树方法,新手入门推荐生物进化树的构建目录前言 (2)一、 NCBI (6)二、 Mega (9)三、 DNAMAN (15)四、DNAStar (18)五、 Bio edit (21)前言1.背景资料进化树(evolutionary tree)又名系统树(phylogenetie tree)进化树,用来表示物种间亲缘关系远近的树状结构图。
在进化树中,各个分类单元(物种)依据进化关系的远近,被安放在树状图表上的不同位置。
所以,进化树简单地表示生物的进化历程和亲缘关系。
已发展成为多学科(包括生命科学中的进化论、遗传学、分类学、分子生物学、生物化学、生物物理学和生态学,又包括数学中的概率统计、图论、计算机科学和群论)交叉形成的一个边缘领域。
归纳总结生物进化的总趋势有以下几类:①结构上:由简单到复杂②生活环境上:由水生到陆生③进化水平上:由低等到高等一般来说,进化树是一个二叉树。
它由很多的分支和节点构成。
根据位置的不同,进化树的节点分为外部节点和内部节点,外部节点就是我们要进行分类的分类单元(物种)。
而物种之间的进化关系则用节点之间的连线表示。
内部节点表示进化事件发生的地方,或表示分类单元进化的祖先。
在同一个进化树中,分类单元的选择应当标准一致。
进化树上不同节点之间的连线称为分支,其中有一端与叶子节点相连的分支称为外枝,不与叶子节点相连的分支称为内枝。
进化树一般有两种:有根树和无根树。
有根树有一个鲜明的特征,那就是它有一个唯一的根节点。
这个根节点可以理解为所有其他节点的共同祖先。
所以,有根树能可以准确地反映各个物种的进化顺序,从根节点进化到任何其他节点只有能有一条惟一的路径。
无根树则不能直接给出根节点,无根树只反映各个不同节点之间的进化关系的远近,没有物种如何进化的过程。
但是,我们可以在无根树种指派根节点,从而找出各个物种的进化路径。
无根树有根树放射树分子进化树(以分子数据为依据构建的进化树)不仅精确地反映物种间或群体间在进化过程中发生的极微细的遗传变异(小至一个氨基酸或一个核昔酸差异),而且借助化石提供的大分子类群的分化年代能定量地估计出物种间或群体间的分化年代,这对进化论的研究而言无疑是一场革命。




系统进化树的这些知识,你都Get了吗?系统进化树(Phylogenetic tree,又称为系统发生树/系统发育树/系统演化树/进化树等),是用来表示物种间亲缘关系远近的树状结构图。
在系统进化树中,物种按照亲缘关系远近被安放在树状结构的不同位置,因而,进化树可以简单地表示生物的进化过程和亲缘关系。
自达尔文时期,很多生物学家就希望用一棵树的形式描述地球上所有生命的进化历程。
早期的系统发育研究主要基于生物的表型特征,通过表型比较来研究物种之间的进化关系,然而,利用表型特征进行系统发育分析存在很大的局限性,1965[1]年,Linus Pauling等提出了分子进化理论,基于分子特性(DNA、RNA和蛋白质分子),推断物种之间的系统发生关系,由于核苷酸和氨基酸序列中含有生物进化历史的全部信息,因此利用该方法构建的系统进化树更为准确。
图1 系统进化树理论上,一个DNA序列在物种形成或者基因复制时,会分成两个子序列,因而系统进化树是一般是二叉树,由许多节点和分支构成。
根据位置的不同,节点分为外部节点和内部节点,外部节点代表最终分类,可以是物种、群体,或者DNA、RAN、蛋白质等,内部节点表示该分支可能的祖先节点,不同节点间的连线则称为分支。
根据是否指定根节点,将系统发育树分为有根树和无根树。
有根树绘制过程中需要引入外群,因而具有一个根节点,作为树中所有物种(样本)的共同祖先节点,可以判断演化方向,反映分类单元间的进化关系,外群与进化树中其他物种(样本)的亲缘关系不宜太近,也不能太远,一般构建种内不同品种/亚种间的进化树,外群应选择同属内其他物种,构建属内不同种间的进化树,外群应选择科内其他属物种。
无根树绘制过程中并未引入外群,因而没有根节点,无法判断演化方向,只能表明不同单元之间的分类关系。
图2 无根树[2](左)和有根树[3](右)此外,系统进化树还可以根据分支长度是否具有意义分为标度树和非标度树。
标度树的分支长度表示变化的程度,而非标度树的分支只表示进化关系,支长无意义。
⼀⽂读懂进化树(图⽂详解)⽬录Content⼀、什么是进化树⼆、进化树的构成1. 根 (Root)2. 结点 (Node)3. 进化⽀ (Branch)4. 外群5. 进化分⽀长度6. 距离标尺7. Bootstrap value三、进化树评估1. Bootstrap检验2. 重复取样值3. Bootstrap value 阈值4. Bootstrap value 与分⽀四、⼏种进化树图1.经典树图(Traditional)Rectangle Tree2.圈图(Circle Tree)3.辐射树(Radiation Tree)什么是进化树系统发育进化树 (Phylogenetic tree):⼀般也叫系统进化树,进化树。
它可以利⽤树状分⽀图形来表⽰各物种或基因间的亲缘关系。
建进化树的过程,⽤术语讲:分⽀系统发育分析 (Molecular phylogenetic analysis):是⽤来研究物种或序列进化和系统分类的⼀种⽅法。
⼀般研究对象是碱基序列或氨基酸序列,通过数理统计算法来计算⽣物间进化关系。
最后,根据计算结果,可视化为系统进化树。
进化树的构成我们模拟⼀个项⽬,使⽤⼈和⿏的各两个基因做进化树,结果如下:可以看到上⾯有⼀堆标注,下⾯来看看它们代表什么意义:1. 根 (所有分⽀的共同祖先叫做根根据有⽆根可分为:有根树:上⾯的图就是有根树,可以从树中找到共同的祖先。
⽆根树:顾名思义,没有根,也就找不到共同的祖先。
⽐如后边会提到的 Straight Tree2. 结点 (每个结点代表⼀个分类单元,物种上可以是属,种群等,基因上可以是基因家族,同源物等。
这⾥需要注意,有的⼈会把 node 翻译为节点,但是节点与结点有着不⼀样的含义:节点:通常被认为是⼀个实体,⽐如互联⽹上的每台计算机,蛋⽩互作⽹络的每个蛋⽩质。
结点:只是⼀个交叉点,指交汇点,并不代表⼀个实体或事物但是,也有另外⼀种解释:这种解释将 node 分为外部节点与内部节点:外部节点⼜叫叶节点,也就是最外层的⼈基因1,⼈基因2等,代表参与分析的序列样本内部节点,也就是我们使⽤蓝⾊标注的位置,代表假定祖先。
一、名词解释1.系统发育树(phylogenetic tree,又称evolutionary tree进化树):是描述群体间进化顺序的分支图或树,表示群体间的进化关系。
2.主成分分析(PCA):是指将多指标化为少数几个综合指标的一种统计分析方法,能够反映原始变量的绝大部分信息。
3.群体结构:是指一个群体内部的基因频率在不同子群体之间存在着系统性的差异。
二、填空题1.我们公司现有的两种简化基因组测序技术分别是RAD和dd-GBS。
2.简化基因组的主要应用有SNP标记的开发、遗传图谱的构建、群体遗传学分析和QTL 分析。
3.目前用于做RAD测序数据的SNP calling的软件是Stacks。
4.遗传图谱中的遗传距离用厘摩(cM)来表示,1 cM的大小大致符合1%的重组率。
三、选择题1.在构建遗传图谱的时候,通常推荐样本数量至少在B个以上。
A. 50B. 100C. 150D. 2002.遗传图谱是指基因或者DNA标记在染色体上以A表示相对位置的图。
A. 遗传距离B. 物理距离3.常见的暂时性分离群体有A和B;常见的永久性分离群体有C和D。
A. F2B. BC1C. RILD. DH4.为了达到彼此相当的作图精度,所需的群体大小顺序为A>C>B≈D。
A. F2B. BC1C. RILD. DH5.我们公司目前的测序平台有(多选):A. Hiseq2000B. Hiseq2500C. Hiseq4000四、问答题1.RAD 技术的主要流程包括哪几个方面?抽提DNA,质检,建库,测序2.RAD 技术有什么特点和优势?特点:(1)通过酶切作用对基因组特定区域进行测序;(2)反映部分基因组序列结构(变异)信息。
优势:(1)测序量低,价格便宜;(2)数据利用率高,性价比高;(3)实验操作简单;(4)能够构建高密度的分子图谱;(5)不依赖参考基因组,物种适用范围广。
3.RAD 技术和 dd-GBS 技术的主要区别是什么?dd-GBS 技术不对 DNA 片段打断,不需要挖胶和纯化,实验周期比较短。
步步图解iTol-给进化树做个美颜系统发育进化树(Phylogenetic tree):一般也叫系统进化树,进化树。
是指以树状结构表示各个节点的进化关系,枝点可以是物种、同一物种的样本、基因等单元。
在细菌基因组和宏基因组(如肠道菌群)的研究中,系统进化树的构建都是生物信息分析中非常重要的一部分。
关于进化树的构建和绘制,目前已经有很多成熟的工具,比如Mega、clustalw等等。
构树工具和参数的选择是一个非常复杂且长的故事了,今天这篇小文暂时不介绍。
今天笔者想用多图实操的方式和大家分享一个超级棒的工具iT ol,让自己的进化树能够表达更多的信息。
iTol网址:/01美化第一步:准备进化树nwk格式的输入文件nwk文件可以通过多种软件获得,例如MEGA。
为了演示,我们这里提供一个含有5个样本的测试文件。
•(A,((B,C),(D,E))))在iTol网站(/upload.cgi)点击upload上传该nwk文件后,获得基本的tree图。
02美化第二步:给进化分支增加颜色。
假设5个样本中,B和C为1组,D和E为另一组,我们希望通过颜色标注分组情况。
此时,我们可以准备如下文档••••••••••••TREE_COLORSSEPARATOR TABDATAA range #a1d8b1 B range #edfcc2C range #edfcc2Drange #f88aafE range #f88aaf# 注意DATA后面为需要我们根据自己的数据调整的部分。
# 每一行为一行样本# 第一列为样本名称(nwk 文件里的名称),第三列为颜色代码,可以根据自己的喜好随意修改。
将建立的文件的拖拽到iT ol的进化树上,我们可以看到此时的tree已经拥有了色彩。
03美化第三步:增加色条假设A和B采样于同一个地区,C、D和E样本来源于另一个样本,我们希望在进化树上利用色条的方式标注。
此时,我们可以按如下格式新建文件,并拖拽到进化树上。