05多序列比对和进化树分析
- 格式:ppt
- 大小:10.35 MB
- 文档页数:58




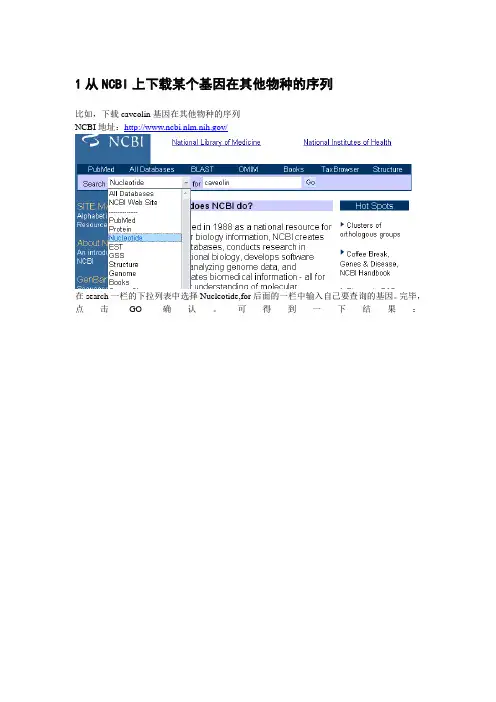
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。

渐进法的策略I.将序列两两比对II.根据相似值将序列分组III.进行组间比对,并继续分组,直至取得最终结果Principle:比对过程中,相似性高的序列先比对,距离远的序列添加其后值与分歧时间t呈非线性关系,原因之一:多个氨基酸替代出现在同一位点。
基于泊松分布对p进行校正,得两序列间每位paralogsorthologs paralogs orthologsErik L.L. Sonnhammer Orthology,paralogy and proposedand proposed classification for paralog subtypes TRENDS in Genetics Vol.18 No.12 December 2002UPGMA方法例:OTU1和OTU2都是原始类群,n1=1,n2=1 OTU r1含两个原始类群OTU1和OTU2 ,nr1=2,OTU3是原始类群,n3=1简明生物信息学,钟扬等主编,用UPGMA法构建的系统树常用构树法比较/phylip/s oftware.htmlHere are 386phylogeny packages and 52free servers, all that I know about. It is an attempt to be completely comprehensive. I have not made any attempt to exclude programs that do not meet some standard of quality or importance….Many of the programs in these pages are available on the web, and some of the older ones are also available from ftp server machines.。


生物信息学中的序列比对与序列分析研究序列比对与序列分析是生物信息学领域中非常重要的研究内容之一。
在基因组学和蛋白质组学的快速发展下,对生物序列的比对和分析需求不断增长。
本文将介绍序列比对和序列分析的概念、方法和应用,并探讨其在生物学研究中的重要性。
一、序列比对的概念与方法:1. 序列比对的概念:序列比对是将两个或多个生物序列进行对比,确定它们之间的相似性和差异性的过程。
在生物信息学中,序列通常是DNA、RNA或蛋白质的一连串碱基或氨基酸。
序列比对可以用来寻找相似性,例如发现新的基因家族、识别保守的结构域或区分不同的物种。
2. 序列比对的方法:序列比对的方法可以分为两大类:全局比对和局部比对。
全局比对将整个序列进行比对,用于高度相似的序列。
而局部比对则将两个序列的某个片段进行比对,用于相对较低的相似性。
最常用的序列比对算法是Smith-Waterman算法和Needleman-Wunsch算法。
Smith-Waterman算法是一种动态规划算法,它在考虑不同区域的匹配得分时,考虑到了负分数,适用于寻找局部相似性。
而Needleman-Wunsch算法是一种全局比对算法,通过动态规划计算最佳匹配得分和最佳比对方式。
二、序列比对在生物学研究中的应用:1. 基因组比对:序列比对在基因组学中具有广泛的应用。
它可以帮助研究人员对特定基因进行鉴定,发现重要的调控元件以及揭示物种间的基因结构和功能差异。
此外,基因组比对还可以用于揭示突变引起的遗传疾病和肿瘤等疾病的发病机制。
2. 蛋白质结构预测:序列比对在蛋白质结构预测中也起着重要的作用。
通过将待预测蛋白质序列与已知结构的蛋白质序列进行比对,可以预测其二级和三级结构以及可能的功能区域。
这些预测结果对于理解蛋白质的功能和相互作用至关重要。
3. 分子进化分析:序列比对在分子进化研究中也扮演着重要的角色。
通过将源自不同物种的基因或蛋白质序列进行比对,可以构建进化树,研究物种的亲缘关系和演化历史。
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。


一、实验目的1. 掌握多序列比对的基本原理和方法。
2. 熟悉使用BLAST、CLUSTAL W等工具进行多序列比对。
3. 分析比对结果,了解序列间的进化关系。
二、实验原理多序列比对是指将两个或多个生物序列进行排列,以揭示序列间的相似性和进化关系。
通过比对,可以识别保守区域、功能域和结构域,为生物信息学研究和进化生物学研究提供重要依据。
多序列比对的方法主要包括以下几种:1. 动态规划法:通过构建一个动态规划表,计算最优比对路径,实现序列的比对。
2. 人工比对法:通过分析序列结构、功能域等信息,人工进行比对。
3. 基于启发式算法的比对:通过寻找序列间的相似性,快速进行比对。
三、实验材料1. 仿刺参EGFR基因氨基酸序列(Fasta格式)。
2. 同源序列数据库(如NCBI)。
3. 多序列比对软件(如BLAST、CLUSTAL W)。
四、实验步骤1. 使用BLAST工具进行同源序列搜索。
(1)在NCBI网站上,选择“BLAST”功能。
(2)将仿刺参EGFR基因氨基酸序列粘贴到“Query Sequence”框中。
(3)选择合适的比对参数,如“MegaBLAST”。
(4)点击“BLAST”按钮,等待结果。
(5)在结果页面,找到相似度最高的几个序列,下载下来。
2. 使用CLUSTAL W进行多序列比对。
(1)将下载的同源序列整合到一个Fasta格式的文本文件中。
(2)在CLUSTAL W软件中,选择“Multiple Sequence Alignment”功能。
(3)上传Fasta格式的文本文件。
(4)选择合适的比对参数,如“Gap Penalty”和“Gap Reward”。
(5)点击“Align”按钮,等待结果。
3. 分析比对结果。
(1)观察比对结果,分析序列间的相似性和进化关系。
(2)绘制系统进化树,展示序列的进化历程。
五、实验结果与分析1. 使用BLAST工具,找到与仿刺参EGFR基因氨基酸序列相似度最高的几个序列,如Anopheles gambiae、Nasonia vitripennis等。
比如,下载caveolin基因在其他物种的序列NCBI地址:在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
快速修剪-多序列比对结果-构建靠谱的进化树展开全文写在前面年初在汕头婚宴上,我问一个朋友,现在在做什么工作。
他回复了我:但行好事,莫问前程。
现在看来,挺好。
构建进化树的基础是序列对齐,或者说多序列比对。
做序列对齐的主要目的是,确定所有序列的同源位点相互对应。
目前存在各种各样的多序列比对算法,但是不存在一个算法能够绝对地保证其能进行完美的位点对应。
此外,我们还需要考虑,我们用于比对的序列可能存在一些错误或者删除和缺失。
所以,一般在我们得到多序列比对结果之后,用于进化树构建之前,我们会多序列比对结果进行修剪。
针对多序列比对修剪,目前存在各种各样的操作,其中包括:1.人工修剪,换句话说,看心情,想删除哪些就删除哪些2.删除所有含有gaps的位点,大体也可以人工删除3.删除不保守位点,比如使用G-blocks等软件4.删除含有一定比例gaps的位点,比如使用MEGA内置的算法5.按照位点信息量进行删除,比如使用trimAL6....基于个人的项目经验,一般我们都直接使用trimAL。
而早前课题组的师弟师妹在做一些家族鉴定工作时,提到能否在界面下(windows/macOS)使用trimAL。
我一直没做回应。
不过我觉得这个事情过于简单。
所以在某一天,我已经将其打包进去。
于是,现在TBtools中存在三个多序列比对结果的修剪逻辑。
1.按照一定比例删除gaps,模仿mega2.删除不保守位点,参考G-blocks的文献,我重新用Java写的3.trimAL,这个软件还在更新,所以我选择直接调用功能界面如下从图片上来看,这三个功能都只是•输入文件或者直接黏贴文本(对于trimAL,我加了自动识别多序列比对格式识别功能,支持的格式很多,包括faslta,clw....)•输出文件或者直接输出文本(对于trimAL,支持多种输出格式,faslta,clw....)•一定的参数控制•隐藏功能,剪切完自动可视化剪切结果输入的多序列比对结果输出的剪切后的结果写在后面并没有太多需要说的。
序列⽐对,构建进化树1从NCBI上下载某个基因在其他物种的序列⽐如,下载caveolin基因在其他物种的序列NCBI地址:/doc/6a02cec358f5f61fb73666e9.html /在search⼀栏的下拉列表中选择Nucleotide,for后⾯的⼀栏中输⼊⾃⼰要查询的基因。
完毕,点击GO确认。
可得到⼀下结果:每⼀条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁⽂的⼈类的字母,表⽰物种,表⽰基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表⽰此序列为cDNA,不含内含⼦。
下图中的NEXT表⽰翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上⾯的这⼀串(碱基)核酸序列。
复制黏贴(包括上⾯表⽰顺序的数字)到TXT⽂本中备⽤。
打开DNAMAN软件,左上⾓点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT⽂本中得)复制到箭头指⽰处,得到:并按照上图左上⾓file-save as(注意此⽂件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存⽅法。
2 序列编辑和⽐对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的⼀部分,所以为了进⾏不同物种间的⽐对,要把下载下来的其他物种的某个基因的序列进⾏删减,以使两段基因是⼤约相同长度的⽚段进⾏⽐对。
以⼈类caveolin1基因为例说明⼀下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进⽐对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要⽐对的序列。
可以按住ctrl点击你要⽐对的⼏个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才⽐对重合部分的后端的碱基数,把这个碱基后⾯的序列删掉,再⽤此⽅法把⽐对重合部分前段得序列删掉,保存。