几种基因_环境基因_基因交互作用研究方法的样本量比较
- 格式:pdf
- 大小:279.53 KB
- 文档页数:4



心理科学进展 2017, Vol. 25, No. 8, 1310–1320 Advances in Psychological ScienceDOI: 10.3724/SP.J.1042.2017.013101310基因与环境的交互作用:来自差别易感性模型的证据*赵德懋 冯姝慧 邢淑芬(首都师范大学心理学系, 北京 100048)摘 要 差别易感性模型认为, 携带某种基因型的个体既容易受到消极环境的不利影响, 同时也容易受到积极的、支持性环境的有利影响。
随着定量遗传学和分子遗传学技术的不断发展, 涌现出关于基因−环境对儿童发展交互作用的大量研究, 主要包括5-HTTLPR 、DRD 4、MAOA 、COMT 和BDNF 五种基因与环境因素(如, 母亲敏感性、压力性生活事件和家庭养育环境等)对儿童发展的交互作用, 支持了差别易感性模型。
但是, 关于基因与环境交互作用的机制、携带易感性基因个体的种族和性别差异问题以及优势敏感性假说的验证, 都是该领域未来研究的重要方向。
关键词 基因−环境交互作用; 基因; 差别易感性; 优势敏感性 分类号B845当前心理学界中一个公认的观点——人类的发展结果不仅仅是由先天生物因素或后天环境因素决定的, 而是两者交互作用的结果, 这一理论思想早已提出, 但是缺乏相应的实证研究。
近年来, 由于定量遗传学和分子遗传学技术的发展, 使得人类在理解基因−环境对个体发展的交互作用方面取得了重大进展(Ellis, Boyce, Belsky, Bakermans-Kranenburg, & van Ijzendoorn, 2011)。
差别易感性模型(Differential Susceptibility Model)是解释基因−环境交互作用的最重要理论之一, 该理论模型巧妙地化解了遗传与环境的争论, 可以合理地解释为什么在相同环境中不同个体会出现不同的发展结果, 为外部环境与儿童发展领域的研究提供了新的范式和方向(Windhorst et al., 2015), 是当前心理学的研究热点和前沿问题之一。

国际医学放射学杂志InternationalJournalofMedicalRadiology2021May 鸦44穴3雪:249-253人脑影像表型的基因组与暴露组广度关联研究于春水△【摘要】脑影像技术可以精确刻画人脑结构与功能,其个体变异决定了人类认知功能及神经精神疾病易感性的个体差异。
人脑结构与功能的个体差异与遗传变异、环境暴露、遗传-遗传交互作用、环境-环境交互作用及遗传-环境交互作用有关,需要从整个基因组和暴露组广度进行系统研究。
重点讨论基因组广度关联研究、暴露组广度关联研究、基因组广度遗传-遗传交互作用研究、暴露组广度环境-环境交互作用研究及基因组-暴露组广度遗传-环境交互作用研究在揭示人脑结构与功能个体差异成因中的潜在价值及其面临的挑战。
【关键词】磁共振成像;基因组;暴露组;人脑中图分类号:R394;R445.2文献标志码:AGenome -and exposome -wide association studies of human brain imaging phenotypes YU Chunshui △.Department of Radiology and Tianjin Key Laboratory of Functional Imaging,Tianjin Medical University General Hospital,Tianjin 300052,China.△E-mail:******************.cn【Abstract 】Human brain structure and function can be precisely characterized by brain imaging techniques and theirinter -individual variations are associated with individual differences in cognitive abilities and susceptibility to neuropsychiatric disorders.Individual differences in human brain structure and function can be attributed to genetic variations,environmental exposures,as well as gene -gene,environment -environment and gene -environment interactions.These effects should be unbiasedly investigated from the whole genome and exposome.Here,we discuss the potential values and challenges in investigating individual differences in brain structure and function by genome -wide association,exposome-wide association,genome-wide gene-gene interaction,exposome-wide environment-environment interaction and genome-exposome-wide gene-environment interaction analyses.【Keywords 】Magnetic resonance imaging;Genome;Exposome;Human brainIntJMedRadiol,2021,44(3):249-253作者单位:天津医科大学总医院医学影像科,天津市功能影像重点实验室,天津300052△E-mail :******************.cn 基金项目:国家重点研发计划“重大慢性非传染性疾病防控研究”重点专项(2018YFC1314300);国家自然科学基金重点项目(82030053)DOI:10.19300/j.2021.S18881专家述评以MRI 为代表的脑影像技术可以准确评估人脑结构与功能,其个体变异决定了人类认知功能及神经精神疾病易感性的个体差异[1]。

基因环境交互作用的统计学研究方法基因环境交互作用是指基因和环境之间相互影响的现象,其中基因和环境的作用不是简单的加法,而是一种相互作用的关系。
基因环境交互作用对个体的发育和进化具有重要作用。
因此,为了研究基因环境交互作用的统计学方法,已成为现代遗传学和生态学的重要研究方向之一。
基因环境交互作用的研究需要考虑两个要素:遗传变异和环境变异。
遗传变异指不同个体之间的基因差异,而环境变异则包括不同个体之间或同一个体在不同环境条件下的表现差异。
这两个要素的交互影响导致了基因环境交互作用的现象。
在现代生物技术的帮助下,研究基因环境交互作用的方法不断发展。
统计学方法在生物研究中有着广泛的应用,也成为研究基因环境交互作用的重要工具。
下面将详细介绍几种常用的统计学研究方法。
1. 方差分析方差分析(ANalysis of VAriance,ANVOA)是研究基因环境交互作用的常用方法之一。
它通过比较不同处理组之间的方差大小来分析基因和环境之间的关系。
方差分析可通过单因素方差分析和双因素方差分析进行。
单因素方差分析是研究基因或环境对表现型影响的一种方法。
例如,我们想研究不同基因型对某一性状的影响,就可以将不同基因型的个体分成不同处理组,并进行方差分析。
如果不同处理组间的方差显著,则说明基因型对性状有影响。
双因素方差分析则是同时分析基因和环境对表现型影响的方法。
例如,我们想研究不同基因型在不同环境条件下对某一性状的影响,就可以将不同基因型的个体分成不同处理组,然后在不同环境条件下进行方差分析。
如果不同处理组间的方差显著,则说明基因环境交互作用存在。
2. 回归分析回归分析是一种寻找变量间关系的方法,它可以用来研究基因和环境间的交互作用。
回归分析可分为线性回归和非线性回归两类。
线性回归是一种用来寻找变量间线性关系的方法,它可以用来研究基因环境交互作用对表现型的影响。
例如,我们想研究不同基因型和环境条件对某一性状的影响,就可以利用线性回归进行分析。

叉生分析在基因一环境交互作用叉生分析是一种方法,用于研究基因与环境之间的交互作用对个体特征或疾病风险的影响。
通过比较同卵双生子(基因相同)和异卵双生子(基因相似度约为50%)之间的差异,可以确定基因对特定环境因素的敏感性,以及基因与环境的相互作用。
叉生分析的基本原理是通过比较同卵双生子和异卵双生子的相似性和差异性。
同卵双生子的基因组是完全一样的,而异卵双生子的基因组则与兄弟姐妹更为相似。
因此,如果在同卵双生子中其中一特征或疾病的发生率更高,那么很可能这是由于不同环境条件导致的。
而如果在同卵双生子和异卵双生子中差异显著,那么研究者有理由相信这是基因与环境交互作用的结果。
在进行叉生分析之前,研究人员首先需要建立双生子队列,收集同卵双生子和异卵双生子的临床数据、生物标本以及环境因素数据。
然后研究人员通过统计分析,计算基因与环境对特定表型的贡献。
叉生分析可以应用在许多领域,包括研究常见疾病(如心血管疾病、糖尿病、癌症等)、复杂疾病(如精神疾病、自闭症、阿尔茨海默病等)以及个体特征(如身高、体重、智力等)。
通过叉生分析,研究人员可以准确地确定基因与环境之间的相互作用,从而更好地理解复杂性疾病的发病机制。
例如,许多研究使用叉生分析来研究肥胖症的发病机制。
通过比较肥胖同卵双生子与肥胖异卵双生子以及瘦卵双生子之间的差异,研究人员可以确定哪些基因变异在特定环境因素下与肥胖的风险相关。
这些环境因素可能包括饮食习惯、运动水平、社会经济地位等。
通过叉生分析,研究人员可以更好地理解个体肥胖风险的遗传和环境基础,以及如何针对不同基因型和环境条件个体定制个体化的干预措施。
叉生分析的局限性包括研究样本的选择偏差、统计算法的复杂性以及环境因素的测量和分类问题。
另外,基因与环境交互作用的研究需要更大的样本量和更复杂的统计模型。
因此,在应用叉生分析时需要充分考虑这些因素。
总之,叉生分析是一种有效的方法,可以帮助我们研究基因与环境交互作用对个体特征或疾病风险的影响。
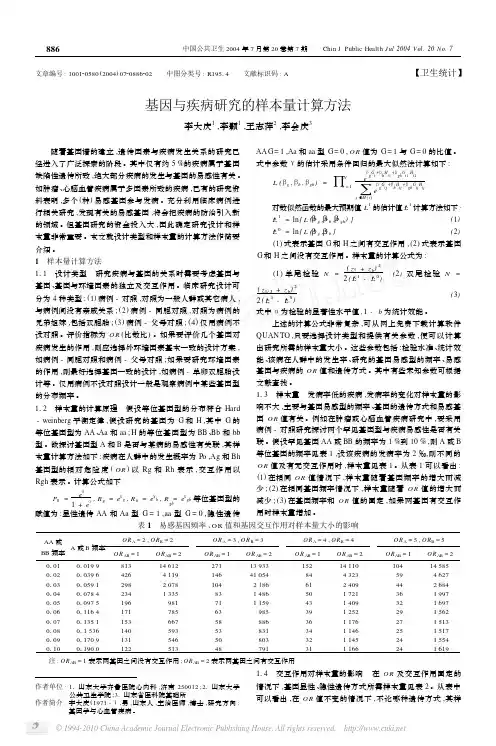
文章编号:100120580(2004)0720886202 中图分类号:R19514 文献标识码:A【卫生统计】基因与疾病研究的样本量计算方法李大庆1,李颢1,王志萍2,李会庆3 随着基因谱的建立,遗传因素与疾病发生关系的研究已经进入了广泛探索的阶段。
其中仅有约5%的疾病属于基因缺陷性遗传所致,绝大部分疾病的发生与基因的易感性有关。
如肿瘤、心脑血管疾病属于多因素所致的疾病,已有的研究资料表明,多个(种)易感基因参与发病。
充分利用临床病例进行相关研究,发现有关的易感基因,将会把疾病的防治引入新的领域。
但基因研究的资金投入大,因此确定研究设计和样本量非常重要。
本文就设计类型和样本量的计算方法作简要介绍。
1 样本量计算方法111 设计类型 研究疾病与基因的关系时需要考虑基因与基因、基因与环境因素的独立及交互作用。
临床研究设计可分为4种类型:(1)病例-对照,对照为一般人群或其它病人,与病例间没有亲戚关系;(2)病例-同胞对照,对照为病例的兄弟姐妹,包括双胞胎;(3)病例-父母对照;(4)仅用病例不设对照。
评价指标为OR(比数比)。
如果要评价几个基因对疾病发生的作用,则应选择外环境因素基本一致的设计方案,如病例-同胞对照和病例-父母对照;如果要研究环境因素的作用,则最好选择基因一致的设计,如病例-单卵双胞胎设计等。
仅用病例不设对照设计一般是观察病例中某些基因型的分布频率。
112 样本量的计算原理 假设等位基因型的分布符合Hard-weinberg平衡定律,假设研究的基因为G和H,其中G的等位基因型为AA、Aa和aa;H的等位基因型为BB、Bb和bb型。
欲探讨基因型A和B是否与某病的易感性有关联,其样本量计算方法如下:疾病在人群中的发生概率为Po,Ag和Bh基因型的相对危险度(OR)以Rg和Rh表示,交互作用以Rgh表示。
计算公式如下P0=eα1+eα,R g=eγg,R h=eγh,Rgh=eγgh等位基因型的赋值为:显性遗传AA和Aa型G=1,aa型G=0,隐性遗传AA G=1,Aa和aa型G=0,OR值为G=1与G=0的比值。

垦堕垦堂童生皇堕塑查垫!!竺!旦第42卷第1期Int J Med Parasit Dis,January 2015,V01.42,No.1 ·49··综述·基因一环境交互作用分析方法在流行病学研究中的应用何健杨坤+【摘要】基因一环境交互作用(gene.environment interac ti on)分析方法适合探讨流行病学中遗传和环境多种因素对于疾病发生或传播的作用,并逐渐成为流行病学研究的重要研究手段。
该文从基因.环境交互作用方法的研究设计和统计学方法两方面,综述此方法的最新国内外研究进展.期望拓展在寄生虫病等研究领域的应用。
【关键词1基因.环境交互作用;流行病学;统计学分析Applic ation of ge ne·en vi ro nm en t in t er a c ti o n an a l ys i s in epide miol ogica l st udie s He Ji an,Ya ngKun*.Jiangsu Institute of Parasitic Di s e a s e s,K e y L a b o r a t o r y of Parasitic D i se a s e Control a nd Prevention,Min蠡try of Heal th,J ia rt gsu Provincial Key Laboratory ofParasite Molecular Biolog y,Wu xi 214064,C h i na+Corresponding author:Yang Kun,Em ail:ji pdy k@163.comSu pp on ed b y N a t i o n a l N a t u r a l S c i en c e F o u n d a t i o n of China(81101275)【Abstract】Gen e.en vironm ent interaction anal ysi s,as me tho d of m uh i.fa ct or s tu dv,c an explore the roleof genetic and envi ronment al factors in the prevalenc e an d transmission of di s e a se s,a n d it has be c o meveryi m p or t a n t m eans of ep i d e m i o l o g i c al s t u d y.R e s e a r c h d e s i g n a n d statistical met hods of g e n e—e n v i ro n m e nt inter-action metho d were revie we d in this paper,aiming at pro vi di n g re f er en c e in the flied of parasitic d i s e a se ss tu d y.【Key words】Gene-environment inte ra ct io n;E pi dem io lo gy;St at is tic al a na ly si s随着人们对于疾病发生认识的改观,发现诸互作用分析方法的进展,以期为流行病学病因研如癌症[I之]、慢性病[3-4]、精神疾病[5-6]、媒介传播究提供参考。

中国人群胰腺癌遗传易感性研究胰腺癌目前仍是预后差,死亡率极高的消化道恶性肿瘤,5年生存率小于5%。
2008年,世界范围内估计新发病例为27万多例,估计死亡病例为26万多例,居全球恶性肿瘤死因的第8位。
近年来,全求胰腺癌的发病率已趋平稳,而我国则呈上升趋势,从70年代相对少见到2005年居常见恶性肿瘤死因的第7位。
胰腺癌高病死率的现状一直没得到改善的重要原因是由于病因尚未清楚。
目前手术切除是唯一可能的治愈方法,由于缺乏有效的早期诊断方法,多数病例确诊时已是晚期,丧失手术切除机会。
恶性肿瘤是一种慢性复杂性疾病,是环境和遗传因素长期共同作用的结果。
胰腺癌的家族聚集性提示遗传因素在胰腺癌的发生过程中扮演重要角色。
因此,开展胰腺癌的遗传易感性研究,寻找特异敏感的生物标志物应用于早期诊断,评估疗效及预后,以及应用发展新的治疗靶,对于我国胰腺癌的综合防治具有重要意义。
第一部分SWI/SNF复合物的遗传变异与胰腺癌发生风险关系研究背景:基因的异常转录是癌症发生的基础,染色质高度紧密的折叠阻止了转录因子、辅因子与DNA的结合,所以凡涉及DNA的反应都要克服染色质的高度紧密性。
ATP依赖的染色质重构复合物是利用水解ATP获得的能量来动员核小体,改变染色质的结构,使得转录因子与DNA结合,从而调节靶基因的转录。
其中交配型转换/蔗糖不发酵复合物(mating type switch/sucrosenon-fermenting, SWI/SNF)蛋白家族广泛参与细胞的分化、增殖和DNA修复,作为肿瘤抑制基因被研究得最为深入,大量的研究发现在包括胰腺癌在内的很多人类癌症中都发现SWI/SNF复合物亚基的突变或表达异常。
研究目的:分析SWI/SNF 复合物遗传变异与中国汉族人群胰腺癌发生的关系;评价SWI/SNF复合物关键基因的遗传变异基因-基因,基因-环境交互作用在于在胰腺癌发生过程中的效应。
研究方法:采用两阶段病例-对照研究系统地分析ATP依赖的染色质重构复合物SWI/SNF基因的遗传变异与中国人群胰腺癌发生风险关系。

![一种基于大样本的基因-环境互作关联分析方法[发明专利]](https://uimg.taocdn.com/271293c918e8b8f67c1cfad6195f312b3169ebe7.webp)
专利名称:一种基于大样本的基因-环境互作关联分析方法专利类型:发明专利
发明人:任文龙,肖静,何书
申请号:CN202210394490.2
申请日:20220414
公开号:CN114678066A
公开日:
20220628
专利内容由知识产权出版社提供
摘要:本发明公开了一种基于大样本的基因‑环境互作关联分析方法,通过采集目标个体的基因序列、环境信息、表型信息,构建基因‑环境检测线性混合模型,并基于预处理共轭梯度法、矩估计方法对基因‑环境检测线性混合模型进行求解,构建服从于卡方分布的基因‑环境互作得分测试统计量,然后应用基因‑环境互作得分测试统计量,完成对基因序列与环境信息是否存在互作效应的判断,该方法可以快速识别多个环境信息对基因‑环境互作位点的基因的影响,适用于大样本的基因‑环境互作关联快速检测。
申请人:南通大学
地址:226001 江苏省南通市崇川区啬园路9号
国籍:CN
代理机构:南京经纬专利商标代理有限公司
代理人:田凌涛
更多信息请下载全文后查看。
基因-基因(环境)交互作用分析方法的比较袁芳;刘盼盼;徐进;费丽娟;郝玲妹;邱旭君;张莉娜【摘要】Three methods of analyzing the gene-gene (environment) interactions are compared in the etiology research of complex diseases to analyze their applicability conditions and the advantages and disadvantages. It shows that crossover analysis is simple and easy to apply, but only applicable for analyzing interactions of single genetic factor and single environment factor. Logistic regression is straight-forward in explaining the epidemiological significance of interaction and performs well in analyzing the main effects, but still has limitations in analyzing higher order interactions. Multifactor dimensionality reduction renders a model-free method and is sensitive to high dimensional data, but it is short of accuracy in estimating the main effects. Considering the advantages identified with each of these three methods in analyzing interaction, the author makes some efforts in this paper to integrate these methods aiming at improving effectiveness interaction analysis.%对复杂疾病病因研究中基因一基因(环境)交互作用的3种分析方法进行了比较,剖析了它们的适用条件和优缺点.结果表明:叉生分析简单易行,但只适用于分析单个遗传和单个环境因素的交互作用;Logistic回归易解释交互作用的流行病学意义且能很好地分析主效应,但在分析高阶交互作用时存在局限性;多因子降维法无需指定特定的遗传模式且对高维数据敏感,但无法估计主效应.鉴于这3种方法在分析交互作用时各有其优点,三者联合应用于交互作用分析效果更佳.【期刊名称】《宁波大学学报(理工版)》【年(卷),期】2012(025)004【总页数】5页(P115-119)【关键词】叉生分析;Logistic回归;多因子降维法;交互作用【作者】袁芳;刘盼盼;徐进;费丽娟;郝玲妹;邱旭君;张莉娜【作者单位】宁波大学医学院,浙江宁波315211;宁波大学医学院,浙江宁波315211;宁波大学医学院,浙江宁波315211;宁波大学医学院,浙江宁波315211;宁波市第七医院,浙江宁波315200;宁波市第七医院,浙江宁波315200;宁波大学医学院,浙江宁波315211【正文语种】中文【中图分类】R181;R195.1随着疾病遗传学研究的深入开展,人们发现基因对疾病的影响是非常复杂的,很多疾病并非简单的由单一基因影响,许多常见疾病和复杂性状可能是多个基因和环境因素之间交互作用的结果. 目前已有多个研究称上位性(基因-基因交互作用)在常见的复杂疾病的表型与基因型关系中发挥关键作用[1-3]. 因此,正确地分析基因-基因(环境)的交互作用对于复杂疾病的病因探索或寻找易感基因有着重要意义. 目前用于交互作用分析的方法有很多种,最常用的有叉生分析、Logistic回归、多因子降维法. 此外,还有位点交互作用的分析方法: 全交互作用模型(Full Interaction Model,FIM),贝叶斯基因关联映射法(Bayesian Epistasis Association Mapping,BEAM),最大条件嫡概率模型(Maximum Entropy ConditionalProbability Modeling,MECPM)[4]等. 近几年以决策树为基础的非参数统计方法也可用于交互作用分析,包括分类和回归树(Classification and Regression Trees,CART),随机森林(Random Forest,RF)、FlexTree[5].这些方法各有其适用性,在交互作用的检验效能方面也广泛存在争议. 笔者就应用较广泛、理论相对成熟的叉生分析、Logistic回归、多因子降维 3种方法进行了比较,阐述其在交互作用分析中的意义.1 叉生分析在统计学上,Ottman将基因-环境的交互作用定义为环境暴露因素对含有不同基因型人群的疾病影响不同,或者基因型对不同环境暴露条件下人群的疾病影响不同[6]. 叉生分析就是根据这一定义来分析基因-环境交互作用的基本流行病学单元,它主要采用2×4叉生表分析单个基因和单个环境因素的交互作用[7]. 叉生分析可适用于传统的病例对照研究、病例同胞对照研究、病例父母对照研究,也可用于单纯病例研究、不完全病例对照研究和队列研究设计的资料.1.1 2×4叉生表叉生分析中,基因和环境因素均为二分类变量(表 1),基因型和环境暴露因素可能的 4种疾病风险组合可以表示为: R11(同时暴露于环境因素和遗传因素对疾病的效应),R10(仅暴露于环境因素对疾病的效应),R01(仅暴露于遗传因素对疾病的效应),R00(遗传和环境因素均未暴露时的疾病风险). R00作为 R11、R10、R01共同的对照组,其OR=1. 交互作用的存在与否取决于相加或相乘模型的选择,若所研究的交互作用为相加交互作用,当 R11-R01≠R10-R00时认为存在交互作用,若所研究的交互作用为相乘交互作用,则当R11/R01≠R10/ R00时考虑存在交互作用. 表1 基因(G)与环境(E)因素作用2×4叉生分析表G E 病例对照 OR值意义++a b R11=ah/bg G、E联合作用效应+- c d R01=ch/dg G单独作用效应-+e f R10=eh/fg E单独作用效应-- g h R00=1 共同对照在表1的基础上还可以计算单纯病例组、单纯对照组中的OR值,对环境、遗传因素分别分层后的疾病风险信息可从表2中获得. 可以通过公式计算得到,分层之后各层的OR值之比等于基于相乘模型时的 OR值,单纯病例组与单纯对照组的 OR值之比也等于基于相乘模型时的OR值.1.2 交互作用评价指标Rothman等[8]提出了相加模型下交互作用的定量评价指标,包括交互作用指数(Synergy Index,SI)、交互作用超额相对危险度(Relative Excess Risk of Interaction,RERI)、交互作用归因比(Attributable Proportion of Interaction,AP)和纯交互作用归因比(AP*).当SI=1时,说明G、E间无相加交互作用,相互独立; SI>1,G、E间具有正相加交互作用,同时存在时效应增强; SI<1,G、E间具有负相加交互作用,同时存在时效应减低. 效应指数 SI既可用于交互作用的定性分析,又可用于交互作用的定量测量. SI绝对值越大,说明因素间交互作用越强.RERI表示两因素联合作用与其单独作用之和的差值,用于描述归因于交互作用的危险度的大小.RERI的绝对值越大,说明因素间的交互作用越强;如果没有相加模型的交互作用,RERI=0.表示G、E同时存在时,疾病的总危险性中可归因于其交互作用的比例. AP绝对值越大,说明因素间的交互作用越强.表示由G、E引起的疾病效应中归因于两者的交互作用所占的比例.交互作用为相乘模型时也可以用交互作用指数进行评价[9],当SI=1时,表明G、E间无相乘交互作用; SI>1时,G、E间存在正相乘交互作用; SI<1时,G、E间存在负相乘交互作用.叉生表计算直观,不仅能分析基因和环境因素各自的主效应,还能计算相加或相乘模型下交互作用的值. 由于使用了相同的参考组,可以对遗传、环境以及两者之间交互作用的疾病危险性进行比较. 对于病例对照研究,不仅可以计算单纯病例和单纯对照下的OR值,还可以分层估计各暴露因素的疾病相对危险度. 如果对照的选择具有良好的代表性,单纯对照中各暴露因素的OR值有助于评估人群中遗传和环境因素的分布是否独立. 但叉生分析并不适用于所有的关联研究,只能分析单个遗传和单个环境因素的交互作用,并且要求两者均为二分类变量,当研究多个因素或者暴露和结局变量为等级、连续型变量(剂量反应)时,叉生分析则无法应用. 此外,研究多个因素时,各因素之间的交互作用作为极为复杂,不能简单地描述为相加或相乘模型.表2 分层情况下因素作用的分析E+ EG 病例对照病例对照+ a b c d- e f g h 分层 OR值意义单纯病例组 ag/ce 基于相乘模型的交互作用单纯对照组 bh/df 人群中暴露因素相互独立性相乘模型R11/(R01×R10)=adgf/bceh 基于存在相乘交互作用相加模型 R11-(R01+R10-1) 基于存在相加交互作用遗传因素分层1 ad/bc 暴露于G时,E的作用遗传因素分层2 eh/fg 未暴露于G时,E的作用环境因素分层1 af/be 暴露于E时,G的作用环境因素分层2 ch/dg 未暴露于E时,G的作用2 Logistic回归模型Logistic回归是分析分类预测变量与离散性结果之间关系的常用模型,是一种参数估计方法.Logistic回归中交互项回归系数的估计是以存在相乘交互作用为基础,在分析交互作用时引入一个相乘项进入回归模型,通过相乘项回归系数的估计来判断交互作用是否有意义及其作用大小. 适用于病例对照研究、横断面研究、封闭队列研究和临床研究的资料,当变量间存在多重共线性问题时,不宜用Logistic回归.通常 Logistic回归用于分析相乘模型下的交互作用,仅当自变量为二分类变量时可估计相加交互作用,Logistic回归对连续性自变量相加交互作用估计的可行性目前研究还较少. Knol等[10]采用模拟数据及临床实际例子证明了Logistic回归在分析一个二分类变量、一个连续性变量以及两个连续性变量相加交互作用时的可行性,并且运用SI、RERI和AP定量评价交互作用. 由于该研究数据来源和样本量的局限性,Logistic回归是否可以分析连续性变量的相加交互作用还需进一步研究.2.1 Logistic回归模型分析交互作用Logistic回归模型为logit(Π)=β0+β1G+β2E+β3G×E,其中: β0 为常数项,β1、β2、β3 为回归系数,ORG=exp(β1)、ORE=exp(β2)、ORG×E=exp(β3)分别为遗传因素、环境因素及交互作用项的调整优势比,反映了各因素对疾病发生的作用. β3=0时,ORG×E=1,G、E 间无交互作用; β3>1,ORG×E>1,G、E 间存在正交互作用; β3<1,ORG×E<1,G、E 间存在负交互作用.2.2 Logistic回归模型的扩展应用Logistic回归不仅能够估计基因-基因(环境)的交互作用,还能估计各自的主效应. 偏回归系数与调整优势比OR呈指数函数关系,其流行病学意义容易解释. Logistic 回归虽有着其独有的优势,但仍存在一定的局限性. 模型估计的参数个数有限,向模型中引入主效应项时,可能的交互项会呈现指数增长趋势,这时Logistic回归就不适用于处理这种含有多个因素的交互项. Hosmer等[11]建议当模型中纳入P个参数时,P+1≤min(N1,N0)/10时为最好,其中N1为出现阳性结果的例数,N0为阴性结果出现的例数. 此外,Logistic回归对维度较敏感,由于高维空间中数据稀疏或存在例数为 0的交互项,用 Logistic回归进行参数估计时标准误较大,假阳性率较高,难以检测真正有意义的交互作用.在候选基因位点较多,样本量相对较小的情况下,容易导致模型的过度拟合,随着交互作用阶数的增多,这种局限性就更显著.由于Logistic回归在分析高维数据方面的局限性,Park等[12]提出了用惩罚Logistic回归来分析基因-基因(环境)的交互作用. 惩罚 Logistic回归是对Logistic回归模型进行简单的修正,将修正系数λ与 Logistic 回归模型相结合,进行二次方调整.Logistic回归模型的二项分布对数似然函数为:惩罚Logistic 回归模型的函数为其中: λ为正常数,λ / 2 ||β| |2 为二次方惩罚项.运用牛顿迭代和岭回归对回归系数进行估计,λ选择使似然函数最大的值. 进行二次方惩罚后,采用哑变量对研究因素进行编码,变量间的多重共线性不会降低模型的拟合度,同时解除了样本量大小对模型中因素数目的限制. 另外,应用二次方惩罚解决了因样本量较小或考虑高阶交互作用存在时高维数据稀疏以及空格子的出现导致检验效能降低的问题,维持了模型参数估计的稳定性.3 多因子降维法多因子降维法(Multifactor Dimensionality Re-duction,MDR)将多个因子看作一个多因子组合,其中的因子指的是研究的变量(基因型或环境因素),维指的是研究的多因子组合中的因子数(如基因型数目). MDR方法的主要思想是将多位点基因型之间的基因-基因或基因-环境的交互作用转换成一个具有两水平的新变量(高危、低危),从而将高维的结构降低到一维,使高阶交互作用的分析更易实现. MDR方法分析的自变量为独立分类变量,例如基因型和环境因素,结局变量为二分类变量,例如病例或对照(患病或未患病),治疗的有效或无效. MDR是一种非参数统计方法,无需指定特定的遗传模式,适用于平衡的病例对照研究和不一致同胞对研究[13].MDR分析交互作用一般包括以下几个步骤:(1)随机将研究数据分为K等份,以便进行K重交叉验证. 通常将数据分为10等份,9份作为训练数据,构建MDR模型,另外的1份为检验数据,进行交叉验证. (2)从一系列基因或分类环境因素中选择N个需研究的因子,N个研究因子即可代表N个空间维度. (3)在N维列联表中,根据每个因子不同的观察值水平,列出N个因子的多水平组合,然后分别计数每一单元格内的病例、对照例数. 例如有N个SNP,每个有m个基因型,则有Nm种基因型组合. (4)对N维列联表中的每一单元格进行分类标记,若病例对照数之比大于或等于某一阈值(例如 1)则标记为高危,否则为低危. 如果某一单元格中只有病例无对照则标记为高危,反之则为低危. 这样所有的基因型组合都能被分成高危和低危两个水平,从而有效地达到 N维结构降低到单维两水平的目的. (5)依次列出各因子组合的分类误差,选出错分误差最小的模型. (6)通过检测样本的十重交叉验证来估计模型的预测误差. 模型由 9/10的训练样本拟合,通过1/10的检验样本来估计预测误差. 为了减少数据划分造成的偶然误差,十重交叉验证重复进行10次,取10次的平均误差作为预测误差的无偏估计. 最后用预测误差的平均值筛选出最有可能存在交互作用的模型.4 讨论上述3种方法各有其优缺点,应综合考虑各方法的适用性,在实际分析中几种方法结合使用. 叉生分析简单直观,但因其只能分析单个基因和单个环境因素的交互作用而受到限制. 应用于资料分析时,可先用Logistic回归筛选具有统计学意义的变量,再运用叉生分析具体分析每两个变量的交互作用大小及相对危险度. 惩罚Logistic回归不仅能够分析样本量相对较小的高阶交互作用,而且其拟合的模型比传统Logistic回归模型更稳定.并且应用惩罚Logistic回归的逐步选择法可以较准确地将具有交互作用的因素从众多研究变量中检测出来,这是传统Logistic回归模型所不能做到的.MDR在分析各因素、各水平间交互作用时并不考虑主效应,因此当主效应没有统计学意义时,MDR仍可发现高阶交互作用,但是MDR却不能发现主效应. MDR对高阶交互作用敏感,无论是何种类型的高阶交互作用(超相乘模型、相乘模型、超相加模型、相加模型等),MDR均具有较好的统计学检验效能. 而且惩罚Logistic回归模型和传统的Logistic回归模仅适用于相乘模型的交互作用分析.因此,可将MDR和惩罚Logistic回归方法联合使用,优势互补.人类基因组关联研究和高通量基因分型技术的深入发展,产生了大量有待分析的复杂生物信息数据,对于研究基因-基因(环境)交互作用,尤其是高阶交互作用的统计学计算方法是一项重大的挑战. 目前有学者提出,分阶段进行交互作用分析可以克服计算方法的困难[14]. 例如,在第1阶段,采用快速筛选方法(如 Tuning ReliefF)选择SNPs,使整个SNP集减少到一个子集. 第2阶段,采用复杂的模型方法(如MDR)来选择SNP子集中有意义的交互作用项. 第3阶段,用假设检验来检验交互项的统计学意义. 这一方法在实际应用中是否能真正克服计算困难还有待更多的研究来验证. 尽管用于基因-基因(环境)交互作用分析的方法有多种,但目前没有任何一种方法可以适用于所有的情况,因此仍需要探索更为合适的方法.基因组关联研究中,大量SNPs的识别对于交互作用的分析是一项挑战,但同时也为进一步的深入研究提供了机遇,如果能正确合理地运用各种复杂的统计学方法,可以提高交互作用的检验效能.未来应在寻找更普遍适用的交互作用检测方法、交互作用生物学意义解释方面作出更多的努力.参考文献:[1]Thornton-Wells T A,Moore J H,Haines J L. Genetics,statistics and human disease: Analytical retooling for complexity[J]. Trends Genet,2004,20:640-647.[2]Moore J H. The ubiquitous nature of epistasis in determining susceptibility to common human diseases[J].Hum Hered,2003,56:73-82.[3]Sing C F,Stengard J H,Kardia S L. Dynamic relationships between the genome and exposures to environments as causes of common human diseases[J].World Rev Nutr Diet,2004,93:77-91.[4]Chen L,Yu G,Langefeld C D,et al. Comparative analysis of methods for detecting interacting loci[J]. BMC Genomics,2011,12:344.[5]Garcia Magarinos M,Lopez-de-Ullibarri I,Cao R,et al.Evaluating the ability of tree-based methods and logistic regression for the detection of SNP-SNP interaction[J].Annals of Human Genetics,2009,73:360-369. [6]Ottman R. Gene-environment interaction: Definitions and study designs[J]. Prev Med,1996,25:764-770.[7]Botto L D,Khoury M J. Commentary: Facing the challenge of gene-environment interaction: The two-byfour table and beyond[J]. Am J Epidemiol,2001,153:1016-1020.[8]Rothman K J,Greenland S,Lash T L. Modern epidemiology[M]. 3nd. Philadelphia: Lippincott Williams& Wilkins Publishers,2008:71-85.[9]Khoury M J,Flanders W D. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: Case-control studies with no controls[J]. Am J Epidemiol,1996,144:207-213.[10]Knol M J,Van Der Tweel I,Grobbee D E,et al.Estimating interaction on an additive scale between continuous determinants in a logistic regression model[J].International Journal of Epidemiology,2007,36:1111.[11]Hosmer D W,Lemeshow S. Applied logistic regression[M]. 2nd. Hoboken: John Wiley & Sons Publishers,2000:260-263.[12]Park M Y,Hastie T. Penalized logistic regression for detecting geneinteractions[J]. Biostatistics,2008,9:30.[13]Ritchie M D,Hahn L W,Roodi N,et al. Multifactordimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer[J]. Am J Hum Genet,2001,69: 138-147. [14]Yang C,Wan X,He Z,et al. The choice of null distributions for detecting gene-gene interactions in genome-wide association studies[J]. BMC Bioinformatics,2011,12(Suppl):S26.。
基因-环境交互作用研究概况南京医科大学王守林一、概述疾病都是在致病因素的损伤与机体的抗损伤作用下,因机体稳态调节紊乱而发生的异常生命活动过程。
致病因素主要是来自机体内在或外在的一些因素,是引起疾病发生的必不可少的因素,像传染病那样用单一因素来解释其他疾病的发生已不能满足今天人类对致病因素的认识,许多疾病的发生是遗传因素和环境因素综合作用的结果。
复杂疾病一般由多种遗传与环境因素以及它们的相互作用造成的,在人群中比较常见的,如糖尿病、肥胖症、骨质疏松症、高血压、心血管疾病、自身免疫性疾病等。
复杂性疾病和单基因疾病的一个最显著的区别在于,它不依照经典的孟德尔模式遗传。
在复杂性疾病中,很多位点相互作用并且和环境因素一起影响疾病的形成。
疾病的临床表型一般是几种不同的中间表型的复合体,这些中间表型背后有不同的遗传和环境因素的作用。
复杂性状疾病具有以下特点:遗传模式尚未确定、群体遗传异质性强、外显率低、多基因参与、单一基因作用微弱,同时还受一组环境因素的作用。
遗传因素和环境因素对复杂疾病的作用的分析方法有比较成熟和完备的理论和实践基础,但遗传因素与环境因素交互作用研究方法还不是很成熟。
有证据表明,很多复杂疾病(如:肿瘤、肥胖、高血压等)实际是多基因与环境交互作用的共同结果,因此如何正确分析和评价基因和环境的交互作用在复杂疾病病因学上的作用就显得至关重要。
此外,对于认识和消除致病因素,对疾病的预防、诊断和治疗也具有重要意义。
(一)遗传变异—内因遗传变异几乎与所有的疾病发生有关,基因在有序调控机制下的正常表达是健康的基础。
某个或某些基因的突变、缺失或调控障碍,使相应的蛋白质结构或功能发生变异,导致细胞对环境改变的应答反应失常并引起疾病发生。
遗传变异至少部分解释了对暴露于同样环境因素之后的这种患病危险的个体差别。
由遗传基础决定个体患病的危险,称为遗传易感性,而由环境因素决定个体患病的危险,称为获得易感性。
遗传易感性是多基因遗传中使用的一个特定概念,易感性高,患病的可能性就大;易感性低,患病的可能性就小。
三基因拟合 p 值三基因拟合p值是一种常见的生物统计学方法,用于研究基因与疾病之间的关系。
在过去的几十年中,人们已经发现了许多与疾病相关的基因。
然而,单个基因通常只能解释疾病发生的一小部分原因。
因此,研究人员开始尝试将多个基因组合起来,以更好地解释疾病的发生和发展。
在这篇文章中,我们将探讨三基因拟合p值的原理、应用和局限性。
一、三基因拟合p值的原理三基因拟合p值是一种用于评估三个基因与疾病之间关系的统计方法。
这种方法通常用于研究复杂疾病,如癌症、糖尿病、心血管疾病等。
其原理是将三个基因的遗传变异结合起来,评估它们对疾病的贡献。
具体来说,三基因拟合p值是通过比较观察到的数据和预期数据之间的差异来计算的。
预期数据是基于人口统计学数据和三个基因的遗传变异频率计算的,而观察数据是从疾病患者和健康人群中收集的。
在计算三基因拟合p值时,我们需要考虑几个因素。
首先,我们需要确定三个基因的遗传变异类型,例如单核苷酸多态性(SNP)或结构变异。
其次,我们需要确定每个变异的频率。
最后,我们需要确定每个变异与疾病之间的关系。
这些因素将被用于计算每个个体的三基因拟合p值。
如果一个人的p值小于预设的阈值,那么这个人就被认为是与疾病相关的。
二、三基因拟合p值的应用三基因拟合p值是一种广泛应用于生物医学研究中的统计方法。
它可以用于研究复杂疾病的遗传基础,评估基因组的复杂性,并为个性化医疗提供指导。
以下是三基因拟合p值的一些具体应用:1.评估基因组的复杂性研究人员通常使用三基因拟合p值来评估基因组的复杂性。
简单地说,如果一个疾病只与一个基因相关,那么我们可以使用单基因关联研究来研究这个疾病。
但是,如果一个疾病与多个基因相关,那么我们需要使用三基因拟合p值来评估这种复杂性。
2.研究复杂疾病的遗传基础三基因拟合p值可以用于研究复杂疾病的遗传基础。
例如,研究人员可以使用三基因拟合p值来研究糖尿病的遗传基础。
他们可以选择三个与糖尿病相关的基因,计算每个个体的三基因拟合p值,并比较糖尿病患者和健康人群之间的p值分布。