Gaussview软件使用手册
- 格式:doc
- 大小:909.00 KB
- 文档页数:7

Gaussview软件使用手册GaussView是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView是不在高斯软件的计算模块中,而是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译和放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其他的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1.经过优化的分子结构;2.分子轨道3.任何估算密度的电子密度表面;4.静电潜在表面;5.原子价;6.与振动频率相符的一般模型;7.IR,Raman,NMR,VCD以及其他的频谱;8.结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP和BOMD轨道的模拟;9.整体能量的小部分以及其他来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目是按照逻辑结构来排列的,开始于最简单的项目,然后是引用最广泛的项目,最后是更加复杂和不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。


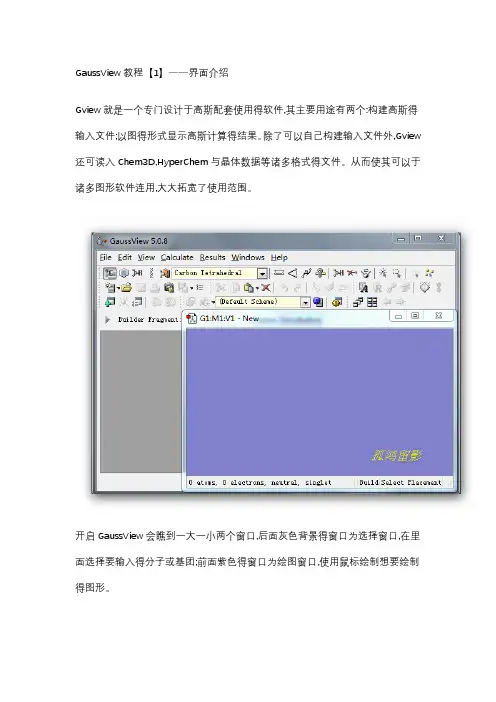
GaussView教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview 还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。

高斯软件基础教程GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏【File】主要功能是建立,打开,保存和打印当前的文件【Edit】完成对分子的剪贴、拷贝、删除、抓图等【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等【Calculate】直接向Gaussian提交计算【Results】接收并显示Gaussian计算后的结果【Windows】控制窗体,如关闭、恢复等【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。

GaussView教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
1、开启Gview2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环在工作窗口单击,可以看到工作窗口也出现了一个苯环。

Gaussview软件使用手册GaussView是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView是不在高斯软件的计算模块中,而是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译和放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其他的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1. 经过优化的分子结构;2. 分子轨道3. 任何估算密度的电子密度表面;4. 静电潜在表面;5. 原子价;6. 与振动频率相符的一般模型;7. IR,Raman,NMR,VCD以及其他的频谱;8. 结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP和BOMD轨道的模拟;9. 整体能量的小部分以及其他来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目是按照逻辑结构来排列的,开始于最简单的项目,然后是引用最广泛的项目,最后是更加复杂和不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。


IntroductionFirst, through its advanced visualization facility, GaussView allows you to rapidly sketch in even very large molecules, then rotate, translate and zoom in on these molecules through simple mouse operations. It can also import standard molecule file formats such as PDB files.Finally, GaussView lets you examine the results of Gaussian calculations using a variety of graphical techniques. Gaussian results that can be viewed graphically include the following:•Optimized molecular structures.•Molecular orbitals.••Electrostatic potential surfaces.•Surfaces for magnetic properties.•Atomic charges.•Animation of the normal modes corresponding to vibrational frequencies.•IR, Raman, NMR, VCD and other spectra.•Animation of geometry optimizations, IRC reaction path following, potential energy surface scans, and ADMP and BOMD trajectories.•Plots of the total energy and other data from the same jobs types as in the previous item.This book provides a reference to all of GaussView's features. Each of the program's features is documented in detail. Items are arranged into several groups based on their general purpose. Within a group, items are arranged in a logical progression, beginning with the simplest, mostIn order to locate the specific information in which you are interested, consult the index for the desired topic or particular dialog. Palettes and dialogs are indexed by their title text (in Bold Type). Menu items are indicated by the form Menu=>Item. Buttons are indexed as "Name button." See the table beginning on page 5 for a list of button names, menu path-button equivalences and keyboard shortcuts.Several hands-on tutorials on various GaussView features are included in the online help. Select the Tutorials topic from the main help screen in order to access them.Previous NextGaussView TutorialsBasic Molecule BuildingMolecule Building Examples and StrategiesThe following examples illustrate how molecules are built in GaussView.Building Pyridine: Using Untyped AtomsThe following is an example of how Pyridine is built in GaussView:1.Start a new file by selecting the New option from the File menu in the View window.2.Select Rings on the Builder window.3.Click on the Active Fragment button. Select benzene from the ring fragments.4.Return to the View window. Click once: benzene displays.5.Click on Element on the Builder window. Click on nitrogen.6.Click on the Active Fragment display. Click on the Nitrogen atom.7.In the View window, click on the carbon atom you want to change to nitrogen.Do not select one of the hybridizations. It is usually easier to use untyped atoms.8.Select the Delete Atom button on the Builder window.9.On the View window, delete the hydrogen attached to the nitrogen by clicking on it.10.Click on the Rebond button on the Builder window, then click on the Clean button.You will often want to rebond before cleaning.Building Phenylpyridine: Setting the Angle Between RingsThis example shows how to build Phenylpyridine based on an existing file, in this case, Pyridine.1.We will be continuing from the previous example, Building Pyridine.2.Click on Rings on the Builder window. Be sure that the phenyl ring is selected.3.Click on the hydrogen atom located on the carbon atom opposite the nitrogen atom on the View window. A second ring is displayed on the screen.4.Select the Dihedral button on the Builder window. Make sure that Move Group is checked.5.Select atoms between the two rings, as shown in the following figureModifying the Dihedral Angle6.Move the slider to create a dihedral until the rings are perpendicular to each other (or enter 90.0 in the text box).7.Click on the OK button.Building Iron Pentacarbonyl: Selecting the Best Templates for a Given TaskThis example builds iron pentacarbonyl.1.Open a new model. Select the element iron and then choose thebi-pyramidal iron template:2.Select the linear C-N template and place this group on each bond to the iron atom. Note that we select this template rather than one of theC-O groups because it is linear.3.Change the nitrogen atoms to oxygen atoms (select the element oxygen and then the free atom template).Building Tyrian Purple Dye: Appending a StructureThe following is an example of how a Tyrian purple dye 6,6'-dibromoindigo is built in GaussView:1. Create a new file.2. Select the fused 5 and 6 carbon rings on the Builder window (see illustration at left) then click on the View window to place a fragment.3.Select the oxygen element and change the appropriate hydrogen atom in the five-member ring to oxygen by clicking on it.4.Change the carbon atom opposite the oxygen to nitrogen.5.Change the hydrogen atom on the outermost carbon atom in the six-member ring (on the same side as the nitrogen) to Bromine in a similar manner.6.S elect Clean on the Builder window. Do not rebond. Then save the file.7.Append the file you just saved to the display on the View window.8.Rotate and move the second ring so that the two carbon atoms to be bonded are close together, and the two fragments are oriented properly with respect to one another.To affect just one of the two fragments, hold down the Alt key while9.Delete the hydrogen atoms on the two carbon atoms to be bonded.10.Select Bond on the Builder window then create a double bond between the two carbon atoms.11.Click on Clean.Docking Two Structures: Building Bichloro-DiphenylThis example illustrates a method for building a molecular structure consisting of docked framents.1.Create a new View window and place a benzene ring in it. Change one of the carbon atoms to Chlorine. Repeat this process to create a second chlorobenzene ring.2.3.Position the rings so that they overlap and the carbon atoms to be bonded are in correct position:4.Delete the hydrogen atoms and dangling bonds in the overlap region:5.ONIOM ExamplesAssigning Atoms to ONIOM Layers in a Small Peptide ChainNote: This example is devised for its illustrative clarity rather than for scientific interest.1.Create a peptide chain containing these amino acids: Phe-Ala-Gly. Choose the Amino-Terminal Phenylalanine, Central Fragment Alanine and Carboxyl-Terminal Glycine.2.Assign all atoms to the Low Layer. Select Assign Layer from the Edit menu, and then click Select All. Verify that current layer is set to Low. The display will look similar to the following figure.Click Apply.3.Assign the following atoms to the Medium layer: all atoms in Ala, carbons in Ring in Phe and atoms up to the double bond in Gly:Add additional atoms (select and then click Apply) as necessary until the selection is correct as in the following figure. Use the center mouse button to add atoms to the selection as needed, and then click Apply to confirm, repeating as needed. The atoms in the Medium layer are nowdisplayed as tubes and the ones in the Low layer are in wireframe in the display:4.Assign the following atoms to the High layer: all atoms in Ala, including heavy atoms bonded to them. Click the central carbon in Ala. Change Using field to Distance. Click twice at the right end of Expand selection. The display will be as in the following figure:Click Apply. The display is as in the following figure:Remove the hydrogen atom that was just placed into the high layer by selecting it and then placing it in the Medium layer.Click Close.Setting Up an ONIOM JobThis example uses the molecule that was built and the layers that were defined in the final example in the Building Tutorial, and it continues on from those steps.1.Select Gaussian from the Calculate menu.2.Click on Multilayer ONIOM Model. Three tabs appear (High, Medium, and Low) in the Method section, which you can use to select the methods for treating each layer of atoms.3.Click on the High tab, then select an RHF/6-31G job.4.Click on the Medium tab, then select a Restricted AM1 job.5.Click on the Low tab and specify a molecular mechanics calculation using the Amber force field.6.The following figure (Gaussian ONIOM Job Setup Example) shows these three tabs in the correct settings (superimposed below one another):These settings place the following line in the job's route section:# ONIOM=(RHF/6-31G:RAM1:Amber)You can now go on to fill out the rest of the dialog box, and the job will be ready to be saved and run.Building Periodic StructuresPeriodic Boundary Conditions (PBC)Tutorial 1 - Build a Trans-Polyacetylene Polymer From Ethylene,PBC/1D1-1Create a new molecule and corresponding view window by clicking on the "New" toolbar button.1-2Make the vinyl fragment (ethylene) the "Current Fragment" bydouble-clicking on the "R-Group Fragment" toolbar button and then clicking on the "vinyl" button in the "Select R-Group Fragment" dialog.1-3Click anywhere in the empty view window to create an ethylene molecule.1-4Center the molecule in the view window by clicking on the "Center" toolbar button.1-5Turn on atom labels for the view window by checking "Labels" from the "View" menu of the main window.1-6In the view window, click on atom 5 of ethylene to create a trans-butadiene molecule.1-7Show the PBC dialog for the molecule by selecting "PBC" from the "Edit" menu of the main window.1-81-9In the "Cell" tab of the PBC dialog, push down the "Place" buttonand make sure "Vertex" is selected to enable placement of the cell origin ("O (0)") vertex at any atom selected in the view window using the mouse. In the view window, click on atom 2.1-10In the "Cell" tab of the PBC dialog, select "a (1)" instead of "O(0)" to enable placement of the "a" cell vertex at any atom selected in the view window using the mouse. In the view window, click on atom 7.1-11In the "Cell" tab of the PBC dialog, select the "Delete All Atoms Outside Cell" item from the "Trim Contents" popup menu.1-12In the "Cell" tab of the PBC dialog, click on the "Center Contents" button.1-13At this point we have defined a primitive unit cell for a PBC/1D model of trans-polyacetylene polymer.1-14In the "View" tab of the PBC dialog, show two cells along the "a"axis using the corresponding spin box in the "Cell Replication" section. By default, the contents of replicate cells are shown in "Low Layer" format.1-15In the "View" tab of the PBC dialog, select "Normal" from the1-16In the "Contents" tab of the PBC dialog, select the "RebondIntercell" item from "Bonds" popup menu to update the bonding between atoms in adjacent cells.1-17experiment. Change the intracell bond length between atoms 1 and 3 by clicking on the "Modify Bond" toolbar button and then clicking on atoms 1 and 3 in the same cell in the view window. In the corresponding "Semichem Smartslide" dialog, change the bond length value from 1.54 to 1.44 in the input field and then click on the OK button.1-18Change the intercell bond length between atoms 1 and 3 by clickingon the "Modify Bond" toolbar button and then clicking on atoms 3 and 1 in adjacent cells in the view window. In the corresponding "Semichem Smartslide" dialog, change the bond length value from 1.38253 to 1.36 in the input field and then click on the OK button.1-19In the In the "Contents" tab of the PBC dialog, select the "RebondAll" item from the "Bonds" popup menu to update the bonding between atoms in the same cell as well as in adjacent cells.1-20Periodic Boundary Conditions (PBC) Tutorial 2 - Build a Graphite Sheet From Benzene, PBC/2D2-1Create a new molecule and corresponding view window by clicking on the "New" toolbar button.2-2Make the benzene fragment the "Current Fragment" by double-clickingon the "Ring Fragment" tool button and then clicking on the "benzene" button in the "Select Ring Fragment" dialog.2-3Click anywhere in the empty view window to create a benzene molecule.2-4Center the molecule in the view window by clicking on the "Center" toolbar button.2-5Turn on atom labels for the view window by checking "Labels" from the "View" menu of the main window.2-6Show the "Point Group" dialog for the molecule by selecting "Point Group" from the "Edit" menu of the main window. In the "Point Group"2-7Change the CC bond lengths to match the experimental values forgraphite. Click on the "Modify Bond" toolbar button and then click on atoms 1 and 2 in the the view window. In the corresponding "Semichem Smartslide" dialog, change the bond length value from 1.39499 to 1.42 in the input field and then click on the OK button. Note that all of the CC bond lengths have increased to 1.42 to preserve the D6h symmetry. In the Point Group dialog, uncheck the "Enable Point Group Symmetry" box and then click the "OK" button.2-8Show the PBC dialog for the molecule by selecting "PBC" from the "Edit" menu of the main window.2-92-10In the "Cell" tab of the PBC dialog, push down the "Place" buttonand make sure the "Vertex" option is selected to enable placement of the cell origin ("O (0,0)") vertex at any atom selected in the view window using the mouse. In the view window, click on atom 6.2-11In the "Cell" tab of the PBC dialog, select "a (1,0)" instead of"O (0,0)" to enable placement of the "a" cell vertex at any atom selected in the view window using the mouse. In the view window, click on atom 2.2-12In the "Cell" tab of the PBC dialog, select "b (0,1)" instead of"a (1,0)" to enable placement of the "b" cell vertex at any atom selected in the view window using the mouse. In the view window, click on atom 4.2-13In the "Cell" tab of the PBC dialog, select the "Delete All Atoms Outside Cell" item from the "Trim Contents" popup menu.2-14Delete the hydrogen atom by clicking on the "Delete Atom" toolbar button and then clicking on the hydrogen atom in the view window.2-15In the "Cell" tab of the PBC dialog, click on the "Center Contents" button.2-16At this point we have defined a primitive unit cell for a PBC/2D model of graphite.2-15In the "View" tab of the PBC dialog, show three cells along boththe "a" and "b" axes using the corresponding spin boxes in the "Cell Replication" section. By default, the contents of replicate unit cells are shown in "Low Layer" format.2-16In the "View" tab of the PBC dialog, select the "Normal" item from2-17In the "Contents" tab of the PBC dialog, select the "Rebond All" item from "Bonds" popup menu to update the bonding between all atoms.2-18Periodic Boundary Conditions (PBC)Tutorial 3 - Build a Primitive Unit Cell of Diamond Crystal FromMethane, PBC/3D3-1Create a new molecule and corresponding view window by clicking on the "New" toolbar button.3-2Make the "Carbon Tetrahedral" fragment the "Current Fragment" by double-clicking on the "Element Fragment" tool button and then clickingon the "Carbon" button and then the "Carbon Tetrahedral" button in the "Select Element Fragment" dialog.3-3Click anywhere in the empty view window to create a methane molecule.3-4Center the molecule in the view window by clicking on the "Center" toolbar button and then position the molecule so all atoms are visible.3-5Turn on atom labels for the view window by checking "Labels" from the "View" menu of the main window.3-6Show the "Point Group" dialog for the molecule by selecting "Point3-7Change the CH bond lengths to match the experimental values for thediamond CC bond length. Click on the "Modify Bond" toolbar button and then click on atoms 1 and 2 in the the view window. In the corresponding "Semichem Smartslide" dialog, change the bond length value from 1.07 to 1.54 in the input field and then click on the OK button. Note that all of the CH bond lengths have increased to 1.54 to preserve the Td symmetry. In the Point Group dialog, uncheck the "Enable Point Group Symmetry" box and then click the "OK" button.3-8Show the PBC dialog for the molecule by selecting "PBC" from the "Edit" menu of the main window.3-93-10In the "Cell" tab of the PBC dialog, push down the "Place" button3-11In the "Cell" tab of the PBC dialog, select "a (1,0,0)" instead3-12In the "Cell" tab of the PBC dialog, select "b (0,1,0)" insteadof "a (1,0,0)" to enable placement of the head of the "b" lattice vector at any atom selected in the view window using the mouse. In the view window, click on atom 4.3-13In the "Cell" tab of the PBC dialog, select "c (0,0,1)" insteadof "b (0,1,0)" to enable placement of the head of the "c" lattice vector at any atom selected in the view window using the mouse. In the view window, click on atom 2.3-14In the "Contents" tab of the PBC dialog, select the "Delete Atoms | Redundant" item from the "Edit" menu of the Atom List editor.3-15In the "Contents" tab of the PBC dialog, change the hydrogen atomto a carbon atom atom by editing the second row of the "Symbol" column in the atom list editor.3-16In the "Contents" tab of the PBC dialog, select "Rebond All" from the "Bonds" popup menu.3-17The bonds between reference unit cells atoms and replicate cell3-18At this point we have defined a primitive unit cell for a PBC/3D model of a diamond crystal.3-19In the "View" tab of the PBC dialog, check the "Show All Boundary Atoms" checkbox to show all atoms in or on the boundary of the cell.3-20In the "View" tab of the PBC dialog, show two cells along each ofthe "a", "b" and "c" axes using the corresponding spin boxes in the "Cell Replication" section.3-21Periodic Boundary Conditions (PBC)Tutorial 4 - Build a Face-Centered Cubic Unit Cell for Diamond Crystal Using Space Group Symmetry, PBC/3D4-1Create a new molecule and corresponding view window by clicking on the "New" toolbar button.4-2Show the PBC dialog for the molecule by selecting "PBC" from the "Edit" menu of the main window.4-34-4In the "Symmetry" tab of the PBC dialog, check the "Enable Space4-5In the "Contents" tab of the PBC dialog, add a carbon atom atfractional coordinates (0, 0, 0) by first selecting "C" from the "Symbol" column and then selecting the "Add" button.4-6length from 4 to the experimental value of 3.56 in the corresponding input field. Note that all of the cell lengths change to 3.56 and also the space group symmetry is preserved because of the "Cell Changes" option being used.4-7In the "Contents" tab of the PBC dialog, select "Rebond All" fromthe "Bonds" popup menu and then select "Dull" instead of "Low Layer" for the "Replicate Format".4-8At this point we have defined a Face-Centered Cubic unit cell for a PBC/3D model of a diamond crystal.4-9In the "View" tab of the PBC dialog, check the "Show All Boundary Atoms" checkbox to show all atoms in or on the boundary of the cell.4-10In the "View" tab of the PBC dialog, show two cells along each of4-11In the "Symmetry" tab of the PBC dialog, uncheck the "Enable Space Group Symmetry" checkbox.4-12Periodic Boundary Conditions (PBC)Tutorial 5 - Transform a Primitive Unit Cell of Diamond Crystal to a Face-Centered Cubic Unit Cell, PBC/3D5-1Follow steps 3-1 through 3-18 of PBC Tutorial 3.5-2Show the PBC dialog for the molecule by selecting "PBC" from the "Edit" menu of the main window.5-35-4In the "View" tab of the PBC dialog, check the "Show All Boundary Atoms" checkbox.5-5In the "View" tab of the PBC dialog, show one replicate cell alongthe "-c" axis using the corresponding spin boxes (1 1 1 0 0 1) in the "Cell Replication" section.5-6In the "Cell" tab of the PBC dialog, select "Fix Contents Cartesian5-7In the "View" tab of the PBC dialog, show zero replicate cells alongthe "-c" axis again, one along the "-a" axis and two along the "+b" axis using the corresponding spin boxes (1 2 1 1 0 0) in the "Cell Replication" section.5-8In the "Cell" tab of the PBC dialog, select "b (0,1,0)" insteadof "a (1,0,0)" to enable placement of the head of the "b" lattice vector at any atom selected in the view window using the mouse. In the view window, click on the atom at position (-1, 2, 0).5-9In the "View" tab of the PBC dialog, show one replicate cell alongthe "+a" axis again, one along the "-b" axis and two along the "+c" axis using the corresponding spin boxes (1 1 2 0 1 0) in the "Cell Replication" section.5-10In the "Cell" tab of the PBC dialog, select "c (0,0,1)" insteadof "b (0,1,0)" to enable placement of the head of the "c" lattice vector at any atom selected in the view window using the mouse. In the view window, click on the atom at position (0, -1, 2).5-11In the "View" tab of the PBC dialog, show just the reference cellusing the corresponding spin boxes (1 1 1 0 0 0) in the "Cell Replication" section.5-12In the "View" tab of the PBC dialog, uncheck the "Show All Boundary Atoms" checkbox.5-13We now have a face-centered cubic unit cell of diamond which givesthe same crystal structure as that from the primitive unit cell we started with.Periodic Boundary Conditions (PBC)Tutorial 6 - Transform a Face-Centered Unit Cell of Diamond Crystal to a Primitive Unit Cell, PBC/3D6-1 F follow steps 4-1 through 4-8 of PBC Tutorial 4.。
Gaussian View使用手册一、引言Gaussian View是一款用于分子建模和计算化学领域的软件,它提供了一系列强大的功能来帮助用户进行分子结构的建模、分析和计算。
本文将为您介绍如何使用Gaussian View软件进行分子建模和计算的基本操作,包括软件的安装、界面的介绍、分子结构的建立和优化、能量和频率的计算等方面的内容。
二、软件安装与启动1. 下载Gaussian View软件安装包,并按照安装向导逐步进行安装。
2. 安装完成后,双击桌面上的Gaussian View图标,打开软件。
三、界面介绍1. 主窗口:软件的主要操作界面,包括菜单栏、工具栏、分子编辑区、控制台等组件。
2. 分子编辑区:用户可以在此处进行分子结构的建立、编辑和优化操作。
3. 控制台:显示软件运行的状态和输出信息。
四、分子结构的建立与编辑1. 新建分子:点击菜单栏中的“File”-“New”选项,选择分子的基本信息,并在分子编辑区中绘制原子和键。
2. 分子编辑:在分子编辑区中可以通过拖动、旋转、缩放等操作改变分子的结构。
3. 分子优化:点击菜单栏中的“Calculate”-“Optimization”选项,对分子结构进行优化。
五、能量和频率的计算1. 能量计算:点击菜单栏中的“Calculate”-“Single Point Energy”选项,对已优化的分子结构进行能量的计算。
2. 频率计算:点击菜单栏中的“Calculate”-“Frequency”选项,对已优化的分子结构进行振动频率的计算。
六、结果分析与导出1. 结果可视化:在控制台中查看计算过程的输出信息和结果,包括能量、振动频率等数据。
2. 结果导出:将分子结构、能量、振动频率等结果导出为文件,方便后续的分析和报告撰写。
七、常见问题与解决方法1. 软件安装问题:如果在安装过程中遇到问题,可以参考官方的安装文档或联系技术支持。
2. 分子结构优化失败:可能是由于初始结构不合理或参数设置不当,可以尝试调整参数或重新建立分子结构。
GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
Gaussview软件使用手册GaussView就是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView就是不在高斯软件的计算模块中,而就是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至就是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译与放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其她的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1.经过优化的分子结构;2.分子轨道3.任何估算密度的电子密度表面;4.静电潜在表面;5.原子价;6.与振动频率相符的一般模型;7.IR,Raman,NMR,VCD以及其她的频谱;8.结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP与BOMD轨道的模拟;9.整体能量的小部分以及其她来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目就是按照逻辑结构来排列的,开始于最简单的项目,然后就是引用最广泛的项目,最后就是更加复杂与不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者就是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。
菜单项目通过菜单=>项目的格式显示出来。
按钮在索引中显示为“名称按钮”。
如下图(从图1开始)显示了一系列的按钮名称,菜单途径—等价于按钮与键盘的快捷键。
在多种GaussView结构的基于指导的帮助包含在了网上帮助中。
在主帮助窗口中选择指导主题来打开她们。
GaussView指导基础分子创建分子创建的例子与策略下面举例说明怎样利用GaussView来创建分子。
创建维生素分子:利用不典型的原子下面就是一个使用GaussView创建维生素分子的例子:1.在View窗口中的File菜单中选择New的选项来创建一个新的文件;2.在Builder窗口中选择好Rings;3.在Active Fragment button上面点击。
在环碎片中选择苯。
4.返回到View窗口。
点击一下演示苯。
5.在Builder窗口中点击Element选项,在氮上点击。
6.点击Active Fragment,点击氮原子。
7.在View窗口中,点击您想变成氮的碳原子。
注意:不要选择杂交原子中的一个,通常选择不典型的原子更加容易。
8.在创建窗口中点击Delete Atom键。
9.在View窗口上,通过点击该键删除与氮相连的氢。
10.在Builder窗口上点击Rebond按钮,然后点击Clean按钮。
注意:通常在清除前应该重新成键。
维生素现在已经完成了。
创建芳环维生素:在环之间设置角度下面的例子展示了怎样在已存在的文件上创建芳环维生素。
在这个例子中,以维生素为基础来创建。
1、您可以接着上述例子来做,创建维生素;2、在Builder窗口上点击Rings按钮。
确保芳环已经被选中;3、在View窗口上点击氮原子对面碳原子上的氢原子。
另一个环就在屏幕上显示了出来;4、在Builder窗口上选择Dihedral按钮。
确保Move Group被标记;5、选择两环之间的原子,如下所示:更改角度6、移动光标来就是的各环所处的平面相互垂直(或者就是在文本框中输入90、0);7、点击Ok按钮;芳环维生素现在完成了。
创建五价铁原子:在指定的项目上选择最好的构型下面就是创建五价铁原子的一个例子1、打开一个新的模型。
选择铁分子然后选择双锥形的铁的构型:2、选择线形的C-N构型然后将该基团连接在铁原子上。
须注意的就是我们选择这种构型而不就是选择一种C-O基团就是因为其就是线形的。
3、将氮原子转换成氧原子(选择氧元素之后再就是自由电子模板)。
现在分子已经完成了。
创建初级紫染:悬挂结构下面就是一个展示怎样在GaussView上面创建一个初级紫染的6,6’-dibromoindigo:1、创创建一个新文件;2、在Builder窗口上选择互联的5与6元碳环(在左侧中的插图中),然后再View窗口上点击来放置一个片段;3、在五元环中,点击合适的氧原子来将其换成氢原子;4、将与氧原子相对的碳原子改变成氮原子;5、将六圆环(在氮原子同一侧的)中的最外层碳原子上的氢原子改成在相同形式的溴原子;6、在Builder窗口上点击Clean按钮。
不要重新成键。
然后保存文件;7、在View窗口上展示刚刚保存的文档;8、旋转与移动另一个环就是的两个碳原子近到足够成键,同时使得两结构方向合适;注意:如果只就是要两个结构中的一个,在按住Alt键的同时拖动鼠标即可。
在展示复杂结构的同时使用另外一个窗口同样也就是很有用处的。
在View菜单上通过Add View选项来创建。
9、将两个成键碳原子上的氢原子删除;10、在Builder窗口上选择Bond来创建一个C=C;11、点击Clean;分子结构完成。
取代两种结构:创建二氯化二苯下面的例子给出了一种包含取代结构的分子结构。
1、创建一个新的View窗口,然后在上面放置一个苯环。
将其中的一个碳原子换成氯原子。
重复该过程创建另一个氯代苯环。
2、旋转其中的一个环,使得氯原子处于另一换的相反位置。
用户可以通过按住Alt键同时按下鼠标左键拖动来完成该过程:3、将环放置使得它们重叠同时碳原子的成键位置正确;4、删除重叠部位的氢原子同时移动键;5、在两个碳原子之间创建一个键,完成分子结构:分子结构完成。
ONIOM例子在一个小的缩氨酸链中的ONIOM层上分配原子注意:这个例子就是为了更好的说明而不就是为了精确1、创建一个包含以下氨基酸的缩氨酸链:Phe-Ala-Gly;选择氨基末端的Phenylalanine,中心Fragment的Alanine以及有羰基末端的甘氨酸。
2、将所有院子分配在最底层,在Edit菜单上选择Assign Layer,然后点击Select All。
确定当前层被设定为Low。
结构大致如下:点击Apply3、将下列原子分配到中间层:所有在Ala中的原子,在Phe环上的碳原子以及甘氨酸双键上的原子:选择Ala中的中心碳原子。
点击Expand Selection的右端然后添加更多的原子(两到三倍)。
添加少量原子通常比添加很多更容易。
点击Apply。
在下列添加附加的原子(选择然后点击Apply)直到结构正确就是十分必要的。
根据需要利用中间的鼠标键来给选项添加原子,然后点击Apply来固定结构,根据需要重复操作。
下图中中间层轨道的原子以棍状出现,底层则以线状出现:4、将下列原子分配到高层上:Ala上所有包括与之相连的重原子在内的所有原子。
点击Ala中的中间碳原子。
将Using场转换成Distance。
在Expand选项的右端点击两次。
结果如下:点击Apply。
然后结果如下:已完成的ONIOM分配的原子点击刚才放置到高层的氢原子然后将其移动(放置)至中间层。
点击Close。
原子层的分配完成。
运行一个ONIOM工作该例子使用了创建的分子,层在创建程序中最终的例子上被定义,然后接着以下步骤来。
1、在Calculate菜单中选择Gaussian。
2、点击Multilayer ONIOM Model。
三种选项(High,Medium与Low)出现在了Method系列中,从这些选项中用户可以分别选择作用于不同层原子的方法。
3、点击High选项,然后选择RHF/6-31G工作。
4、点击Medium选项,然后选择受限制的AM1工作。
5、点击Low选项具体说明一种在使用Amber force区域的原子结构计算方法。
6、下列的结构(高斯ONIOM工作的运行例子)显示了在正常运行状态的着三种选项(一个重叠着一个):这些运行的区域在工作的route section显示:# ONIOM=(RHF/6-31G:RAM1:Amber)用户现在可以继续填写剩下的文本框,然后工作就可以准备好保存并运行了。
创建周期性的结构周期性束缚状态(PBC)指示1——通过乙烯创建一个经过多元炔的聚合体PBC/1D1-1通过点击工具栏的“New”按钮来创建一个新的分子与一个对应的视图窗口。
1-2首先双击“R-Group Fragment”的工具箱按钮,然后点击“Select R-Group Fragment”对话框中的“vinyl”按钮来创建乙烯基(乙烯)(当前的结构)。
1-3在视图窗口中的任意空白位置点击来创建一个乙烯分子。
1-4点击“Center“工具箱按钮来使得视图窗口以该分子为中心。
1-5在住窗口中的“View”菜单中点击“Labels”来打开该视图窗口的原子标签。
1-6在视图窗口中,点击乙烯的原子5来创建一个穿过丁烷基的分子1-7在主窗口的“Edit”菜单中选择“PBC”来显示分子的PBC对话框。
1-8在PBC对话框的“Symmetry”选项中,选择“Lattice Dimensions”多选框中的“1”。
1-9在PBC对话框的“Cell”选项中,在确定“Vertex”被选择的情况下按下“Place”按钮,这样就可以在视图窗口中使用鼠标来选择任何原子的细胞起源定点(“0(0)”)。
在视图窗口中,点击原子2、1-10在PBC对话框的“Cell”选项上,用鼠标在View窗口中点击选择“a(1)”来代替“0(0)”使得a胞的替代。
在View窗口中,点击原子7。
1-11在PBC对话框的Cell选型上,在“Trim Contents”菜单中选择”Delete All Atoms Outside Cell”。
1-12在PBC对话框的”Cell”选项上,点击“Center Contents”按钮。
1-13这样,我们就为穿越多乙炔的聚合体的PBC/1D模型定义了一个初始合成胞体。
1-14在PBC对话框的“View”选型上,使用”Cell Replication”的旋转空间来显示两个沿着“a“轴的胞体。
特殊情况下,折叠胞体的内容会显示在”底层“表格中。
1-15在PBC对话框的“View“选项上,从”Replicate Contents Format”多选框中选择”Normal”在同一个表格中显示出折叠胞体的内容。