Co(Ⅱ)和Ni(Ⅱ)在北山花岗岩上的吸附:表面配位模型及线性自由能关系
- 格式:pdf
- 大小:970.08 KB
- 文档页数:8

U(VI) 在北山花岗岩上的吸附本实验采用批式实验研究了U(VI)在北山花岗岩上的吸附行为,考察了接触时间、离子强度、pH和温度对U(VI)在花岗岩上吸附的影响。
研究结果表明,U(VI)在花岗岩上的吸附会在8h内达到平衡;随着固液比的增大,吸附百分数增大;离子强度对吸附平衡基本没有影响;升高温度促进U(VI)在花岗岩上的吸附。
利用假二级动力学模型对吸附过程进行拟合,数据能够较好的得到吻合。
对吸附等温线进行Langmuir和Freundlich模型拟合,发现实验数据能够较好的符合Langmuir吸附模型。
关键词; 北山花岗岩,U(VI),吸附,温度1.1放射性废物处置核能的和平利用是二十世纪人类最伟大的成就之一。
自1954年前苏联建成了世界上第一座核电机组以来,人类进入了和平利用核能的时代。
与传统能源相比,核电具有能量密度高、输出功率稳定、高效、清洁、低碳、环境友好等特点,在未来的能源结构中具有很强的竞争力。
然而,伴随着核能利用所产生的大量放射性废物,能否安全合理的处理处置这些放射性废物已成为制约核能可持续发展的重要因素。
在这些放射性废物中,高放废物是指放射性核素的含量或浓度高、释热量大、毒性强、半衰期长、操作和运输过程中需要特殊屏蔽的放射性废物,高放废物的体积仅为所有放射性废物的体积的1%左右,但其几乎包含了约90%以上的放射性。
世界各有核国家都将高放废物的安全处置看作是保证核工业可持续发展、保护人类健康和自然环境的一项战略任务。
对放射性废物的处理处置有特殊要求,因此如何安全地处理处置放射性废物已经成为我国乃至世界核能可持续性发展所必须直面的问题。
在众多处置方案中,高放废物地质处置是开发时间最长,也是目前最有希望投入应用的方案。
高放废物地质处置目的是把关键放射性核素保持在特定的地质环境中,确保其在衰变至天然辐射水平的漫长时间中,所释放到生物圈的放射性处于生物可以接受的水平。
高放废物的地质处置一般在地表以下500~1000米具有稳定地质构造的地层中开挖矿山式巷道,以此作为地质处置库的主体。

CO在α-U(001)表面的吸附李赣;罗文华;陈虎翅【摘要】采用密度泛函理论中的广义梯度近似,计算了CO在α-U(001)表面的吸附、解离和扩散.结果表明:CO分子以CU3OU2>构型化学吸附在α-U(001)表面,吸附能为1.78-1.99 eV;吸附后表层U原子向上迁移,伴随着褶皱的产生;CO分子与表面U原子的相互作用主要是U原子的电子向CO分子最低空轨道2π*转移,以及CO2π*/5σ/1π-U 6d轨道间杂化而生成新的化学键:CO解离吸附较分子吸附在能量上更为有利,h1(C)+h2(O)和h1(C)+h1(0)(h:空位)解离态吸附能分别为2.71和3.08 eV;近邻三重穴位之间C、O原子的扩散能垒分别为0.57和0.14 eV,预示O 原子较C原子更易在U(001)表面扩散迁移.【期刊名称】《物理化学学报》【年(卷),期】2010(026)005【总页数】7页(P1378-1384)【关键词】密度泛函理论;α-U(001)表面;CO;吸附【作者】李赣;罗文华;陈虎翅【作者单位】表面物理与化学国家重点实验室,四川,绵阳,621907;表面物理与化学国家重点实验室,四川,绵阳,621907;中国工程物理研究院,四川,绵阳,621900【正文语种】中文【中图分类】O641;O647铀是原子能工业中一种重要核燃料,化学性质十分活泼,在环境气氛中极易发生氧化腐蚀而造成其使用性能发生改变.因此,铀与活性气体的相互作用受到广泛关注.铀的初期氧化一般经历吸附、解离、扩散和氧化物形核长大等阶段,其中,吸附、解离过程是初期氧化物形成的前提.因此,了解活性气体的吸附、解离过程有助于深入理解铀的氧化腐蚀.然而,铀的高化学活性使得吸附、解离作用时间很短,实验上难以观测.因此,需要从理论角度研究活性气体在铀表面的吸附行为.近年来,活性气体在铀表面吸附的理论研究不断见于报道,如Huda[1]和Dholabhai[2]等利用Dmol3程序研究了O2和CO分子在γ-U(001)面吸附的稳定构型、电子结构和磁性质.Nie等[3]利用VASP程序研究了H2分子在α-U(001)面的吸附、解离和扩散过程.Senanayake等[4]在CO与UO2(111)单晶反应的实验工作中,利用PWscf程序简单计算了CO分子在α-U(001)表面的吸附.室温下金属铀以正交结构α相存在,而体心立方结构γ相仅在高温(1050 K)下存在[5],因此,研究活性气体在α-U表面的吸附比γ-U表面更具有代表性.CO分子在α-U表面吸附的理论研究仅见于Senanayake等[4]的工作,但其只是在忽略了表面原子驰豫条件下简单计算了两种典型分子和解离吸附构型,进而指出解离吸附较分子吸附在能量上更加有利,而且对吸附涉及的电荷转移、电子结构和解离活化情况也未作讨论.有鉴于此,本文利用密度泛函理论[6-7]中的广义梯度近似,模拟计算了CO分子在α-U(001)表面的吸附、解离和扩散,较全面地研究了吸附能、吸附构型、相互作用本质、解离和扩散能垒等问题以及吸附对表面U原子结构的影响,以期在原子分子水平上深入认识CO与铀的初期相互作用.从计算效率和计算精度考虑,文中选用Dmol3量子力学程序,模拟计算CO分子在α-U(001)面的吸附.Dmol3采用数值基方法[8-9],U原子内层电子采用包含相对论效应的有效核势(DSPP)予以考虑,价电子波函数采用双数值基加极化函数(DNP)展开,它相当于Gaussian程序中的6-31G**,O原子采用全电子基组;交换关联势采用PW91[10],使用自旋限制波函数求解Kohn-Sham方程;布里渊区积分的Monkhorst-Pack[11]网络参数设为4×3×1,实空间截止半径为0.51 nm;结构优化以能量、位移和力收敛为判据,收敛阈值分别为2×10-5Ha、5×10-4nm和4× 10-2Ha·nm-1.此外,CO分子解离以及解离C、O原子表面扩散过程的过渡态计算采用LST/QST/CG方法[12-13],该方法将线性同步过渡(LST)/四级同步过渡(QST)算法和共轭梯度(CG)方法结合使用,是一种较为有效的过渡态搜索技术.文中全部计算在SGI Altix 350多处理器的工作站上完成.α-U的晶体结构为正交结构(Cmcm),坐标为(0, ±0.102,±0.25)[14],采用p(2×1)的五层U原子层晶(slab)模拟α-U(001)表面(见图1),真空层厚度选取1.5 nm,以确保相邻层晶间的相互作用足够小.图1同时标出了表面4种具有高对称性的吸附位置,其中,A代表顶位,B代表桥位,C代表三重穴位1,D代表三重穴位2.吸附模型构建时,表面上放置一个CO分子,覆盖度为0.25.图2给出了构建的吸附模型示例,对于每一个吸附位,均考虑了三种可能的吸附态,水平吸附(hor1和hor2,CO分子分别平行于X和Y轴)和垂直吸附(ver).一般而言,表面吸附原子的作用范围有限,主要与固体表层和次表层原子发生作用,因此CO/α-U(001)体系构型优化时,底部三层U原子固定,上面两层U原子和CO分子坐标全部放开.吸附能Eads定义为吸附前后体系总能量的变化.其符号和大小可表示吸附发生的可能性和吸附强度.其中ECO/U(001)、EU(001)和ECO分别表示CO/α-U(001)体系、净α-U(001)表面和自由CO分子的能量,Eads值为正时表示吸附体系是稳定的.2.1 方法和模型验证为了验证文中计算方法的可靠性,首先对α-U晶体和CO分子的晶格常数进行了优化.α-U晶体结构参数的计算结果为a=0.2829 nm、b=0.5903 nm、c=0.4928 nm,内坐标y=0.098,与其他理论计算结果[15-17]和实验值(a=0.2836 nm、b=0.5866 nm、c= 0.4935 nm,y=0.102)[14]符合很好,CO分子的优化键长为0.1140 nm,与实验值0.1128 nm[18]的偏差小于1.1%.比对结果说明计算方法是可靠的.其次,分别对构建的3、4、5和6层α-U(001)面层晶模型分别作表层和次表层原子的开放优化,按式(2)计算的相邻第i和第j层之间距离的相对变化百分比Δdij(负号表示收缩,正号表示膨胀)列于表1.式中,dij为弛豫后相邻的第i和第j层之间距;d0为弛豫前理想晶体相邻层之间距.优化结果显示,表面原子几乎没有重构发生,但却有明显的弛豫现象,表层与次表层间距Δd12显著减小,收缩比为2.81%-3.11%,而且Δd12几乎不依赖于模型层数n.次表层与第三层的间距变化显著依赖模型层数,n从3增至4时Δd23显著减小,说明3层模型不能准确描述表面原子的弛豫.n≥5时,Δd23逐渐趋于稳定.计算结果说明文中选取5层层晶模型模拟CO在α-U (001)面的吸附是合适的.2.2 CO分子的吸附2.2.1 吸附构型和吸附能对前述预先构建的12种CO分子吸附模型进行优化,发现仅存在图3所示的4种稳定构型,分别定义为I-IV构型,相应的构型参数和吸附能列于表2.由图3结合表2数据可见,(1)CO分子与U原子以CU3OU2构型的多键方式作用,即C原子与邻近3个U原子键合,C—U距离为0.2191-0.2534 nm,接近于U碳化物晶体中的C—U距离(UC: 0.248 nm;UC2:0.232-0.257 nm;U2C3:0.246-0.276 nm)[19];O与邻近2个U原子键合,O—U键距离为0.2390-0.2583 nm,略大于UO2晶体中的O—U距离(0.237 nm)[20],这说明CO分子与表面U原子具有较强的相互作用.此外,构型I吸附能和C—O键长的优化结果与Senanayake等[4]的计算结果(2.07 eV和0.132 nm)基本一致,说明优化结果对表面原子驰豫并不敏感.(2)CO分子的吸附能为1.78-1.99 eV, C—O距离为0.1296-0.1358 nm,较气态分子的优化值0.1140 nm增大,说明CO在U(001)表面吸附后C—O键削弱,但C—O之间仍存在化学键,即CO分子发生非解离化学吸附,并且从能量上看,CO在α-U(001)表面吸附时,4种吸附构型均有可能存在.此外,结合C—O键与表面的法向夹角(66.9°-78.3°)以及后述的电子转移情况来看,吸附活化的CO可描述为倾斜的COδ-态.(3)与表1所列吸附前的弛豫值相比,吸附后Δd12值增大,增幅为0.69%-1.33%,说明在CO分子作用下,表层U原子向上迁移;而Δd23值变化并不显著(-0.03%-0.15%),说明CO分子主要与表层U原子相互作用,与次表层U原子的相互作用则较弱.此外,源于表层铀原子所受键力的不均质性,表层U原子产生褶皱,褶皱值(Δr)为0.002-0.011 nm.2.2.2 电荷布居和电子结构为了清楚认识CO分子与表层U原子之间的相互作用本质,分别计算了CO/α-U(001)体系的电荷转移和最稳定I-型的态密度.表3给出了CO/α-U(001)体系CO分子的Mulliken电荷分布,图4给出了I-型的态密度(为了比较说明,图中同时给出了净α-U(001)面表层原子和自由CO分子的态密度).图4显示了吸附前自由CO分子的三个最高占据轨道(HOMO)4σ、1π和5σ轨道和一个最低空轨道(LUMO)2π*,以及表面铀原子由于近邻原子数减少而导致的U 5f电子部分局域化(表现为费米能级处U 5f峰较体相U 5f峰锐化)的特征.对比吸附前后体系的态密度可见,(1)表面U原子电子向CO 2π*轨道转移,2π*轨道移至费米能级以下,并与U 5f/U 6d轨道发生杂化增强键合作用;CO 5σ轨道和1π轨道也通过与U 6d轨道发生杂化参与键合,而4σ轨道峰形和峰强基本没有变化,近呈化学惰性.(2)费米能级处U 5f峰强降低,峰形略有展宽,说明少量U 5f电子参与成键.由表3和表2可见,吸附CO分子的C—O键长随其净电荷的增加而增大,这符合CO 2π*反键轨道上得到电子数越多,C—O键削弱程度越大的一般规律.综上可知,CO分子在α-U(001)表面的吸附活化主要是U原子电子向CO 2π*轨道转移,以及CO 2π*/ 5σ/1π-U 6d轨道间杂化而生成新的化学键.2.3 CO分子的解离和表面扩散实验上McLean等[21]利用X射线光电子能谱(XPS)、俄歇电子能谱(AES)等表面分析技术观测到CO在多晶α-U表面解离为C、O原子,随之在表面迁移并向体内扩散的过程,为此本文研究了CO分子在α-U(001)表面的解离以及解离C、O原子在α-U(001)表面的扩散过程.首先计算了孤立C、O原子在α-U(001)表面的吸附,优化结果示于表4.由表4可见,(1)C、O原子在α-U(001)表面均仅存在hollow1 (h1)和hollow2(h2)位两种稳定吸附构型,说明顶位和桥位吸附并不稳定,易向三重穴位1和2转化,这符合原子吸附更易发生于多键位置的一般规律,同时C原子较O 原子距表面U原子更近;(2)由于O的电负性较C原子要大,因此O原子得到的电子数要多,与U原子的结合强度要大,O原子hollow2位的吸附强度略大于hollow1位,而C原子hollow1位的吸附强度略大于hollow2位.其次,由于CO以I-型吸附最为稳定,但次稳定IV-型的C—O键长最大,C—O键的削弱程度最大,因此计算了这2种构型下CO的解离过程,相应的吸附和解离势能曲线示于图5,I-型解离吸附态的态密度示于图6.根据C、O原子的优化结果以及I-型和IV型中CO分子的结构特征,构建的解离产物的优化结果列于表5.由表5可见,解离吸附(2.71和3.08 eV)较分子吸附(1.99和1.95 eV)在能量上更为有利,伴随着表面U原子的向上迁移更加显著.值得注意的是,I-型解离吸附态h1(C)+h2(O)的Δd12和Δr值较IV-型解离吸附态h1(C)+h1(O)的值要大很多,这与C(O)-U原子的作用方式有关,h1(C)+h2(O)吸附态中C、O原子与表层4个U原子均发生作用,这使得U原子的Z向延展性受到协同约束而不易移动;而h1(C)+h1(O)吸附态中C、O原子主要与近邻3个U原子发生作用,这使得O—O间的两个U原子受到较强键力而显著向上迁移.此外,文中优化的h1(C)+h2(O)解离态的吸附能与Senanayake等[4]计算的2.53eV也基本相符,但其未考虑h1(C)+h1(O)解离态,而我们发现该解离态在能量上更为有利.由图5可见,(1)CO分子的解离吸附能较分子吸附能要大许多,且解离势垒(1.02和0.72 eV)较分子吸附能要小许多(若二者数值相当,则解离、脱附过程均可能发生),说明CO分子在一定热激活条件下趋于发生解离.(2)由于IV-型中C—O键削弱程度较大,因此其解离势垒较I-型解离势垒要小.I-型和IV-型解离过渡态中CO的净电荷分别为-1.01e和-1.02e, C—O键长分别为0.1871和0.1870 nm,表明在解离过程中,更多的电子转移到CO分子2π*反键轨道上,使得C—O键得到充分活化,继而断裂分解为C与O原子.由图6结合表5可见,解离C、O原子与表面U原子的相互作用一方面是电子从U原子向C、O原子的2p轨道转移,另一方面是U与C、O原子轨道间的杂化,其中,C 2p与U 5f和U 6d轨道间的杂化发生在费米能级以下0-4.0 eV范围内,而O 2p与U 5f和U 6d 轨道间的杂化发生在费米能级以下4.4-5.6 eV范围内.此外,U 5f电子行为类似于图3所示CO吸附时的情况,这里不再赘述.最后,根据C、O原子的优化结果,分别计算了邻近两个穴位之间C、O原子的扩散:h2(C)→h1(C)和h1(O)→h2(O),相应的最低能量路径(MEP)示于图7.由图7可见,过渡态中C原子位于扩散路径中的桥位,O原子接近扩散路径中的桥位,与近邻两个U原子的键长分别为0.2113和0.2121 nm.计算的h2(C)→h1(C)扩散能垒为0.57 eV,h1(O)→h2(O)扩散能垒为0.14 eV,预示O原子较C原子更易在U(001)表面扩散迁移.采用广义梯度密度泛函理论计算了CO分子在α-U(001)表面上的吸附、解离和扩散.CO分子吸附能和几何构型的优化结果表明,CO分子以CU3OU2构型化学吸附在α-U(001)表面,吸附能为1.78-1.99 eV;吸附后表层U原子向上迁移,伴随着褶皱的产生.电荷布居和态密度的分析表明,CO分子与表面U原子的相互作用主要是U 原子电子向CO 2π*轨道转移,以及CO 2π*/5σ/1π-U 6d轨道间杂化而生成新的化学键.CO解离吸附较分子吸附在能量上更为有利,h1(C)+h2(O)和h1(C)+h1(O)解离态的吸附能分别为2.71和3.08 eV.近邻三重穴位之间C、O原子的扩散能垒分别为0.57和0.14 eV,预示O原子较C原子更易在U(001)表面扩散迁移.研究结果为更好地理解CO与金属铀的初期相互作用提供了一定理论基础.【相关文献】1 Huda,M.N.;Ray,A.K.Int.J.Quantum Chem.,2004,102:982 Dholabhai,P.P.;Ray,pd.,2007,444:3563 Nie,J.L.;Xiao,H.Y.;Zu,X.T.;Fei,G.J.Phys.-Condes.Matter, 2008,20:4450014 Senanayake,S.D.;Soon,A.;Kohlmeyer,A.;Sohnel,T.;Idriss,H.J.Vac.Sci.Technol.A,2005,23:10785 Blanter,M.S.;Glazkov,V.P.;Somenkov,V.A.The Physics of Metal andMetallography,2006,101:1536 Hohenberg,P.;Kohn,W.Phys.Rev.,1964,136:B8647 Kohn,W.;Sham,L.J.Phys.Rev.,1965,140:A11338 Delley,B.J.Chem.Phys.,2000,113:77569 Delley,B.Int.J.Quantum Chem.,1998,69:42310 Perdew,J.P.;Wang,Y.Phys.Rev.B,1992,45:1324411 Monkhorst,H.J.;Pack,J.D.Phys.Rev.B,1976,13:518812 Bell,S.;Crighton,J.S.J.Chem.Phys.,1984,80:246413 Fischer,S;Karplus,M.Chem.Phys.Lett.,1992,194:25214 Barett,C.S.;Mueller,M.H.;Hittermann,R.L.Phys.Rev.,1963, 129:62515 Söderlind,P.Phys.Rev.B,2002,66:08511316 Vitos,L.;Kollar,J.;Skriver,H.L.Phys.Rev.B,1997,55:494717 Nie,J.L.;Xiao,H.Y.;Gao,F.;Zu,pd.,2008, 476:67518 Lide,D.R.CRC handbook of chemistry and physics.Boca Raton: CRC Press,200319 Storms,E.K.;Haber,A.J.J.Nucl.Mater.,1967,23:1920 Wyckoff,R.W.G.Crystal structures 1.New York:Interscience Press,196321 McLean,W.;Colmenares,C.A.;Smith,R.L.Phys.Rev.B,1982, 25:8。

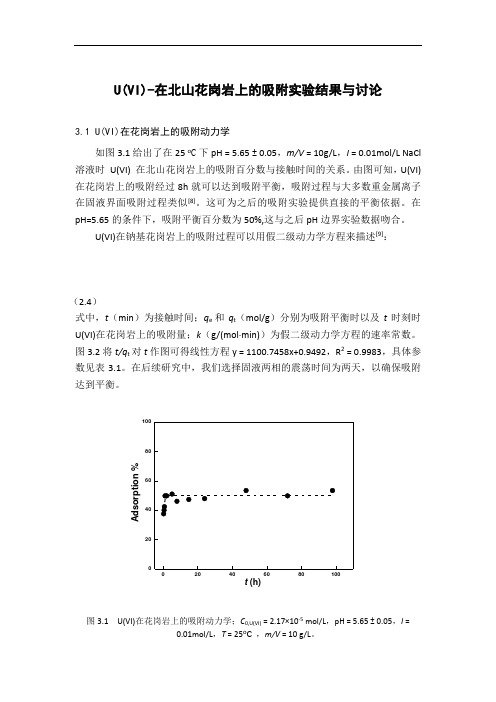
U(VI)-在北山花岗岩上的吸附实验结果与讨论3.1 U(VI)在花岗岩上的吸附动力学如图3.1给出了在25 o C 下pH = 5.65 ± 0.05,m/V = 10g/L ,I = 0.01mol/L NaCl 溶液时 U(VI) 在北山花岗岩上的吸附百分数与接触时间的关系。
由图可知,U(VI)在花岗岩上的吸附经过8h 就可以达到吸附平衡,吸附过程与大多数重金属离子在固液界面吸附过程类似[8]。
这可为之后的吸附实验提供直接的平衡依据。
在pH=5.65的条件下,吸附平衡百分数为50%,这与之后pH 边界实验数据吻合。
U(VI)在钠基花岗岩上的吸附过程可以用假二级动力学方程来描述[9]:(2.4)式中,t (min )为接触时间;q e 和q t (mol/g )分别为吸附平衡时以及t 时刻时U(VI)在花岗岩上的吸附量;k (g/(mol ∙min))为假二级动力学方程的速率常数。
图3.2将t/q t 对t 作图可得线性方程y = 1100.7458x+0.9492,R 2 = 0.9983,具体参数见表3.1。
在后续研究中,我们选择固液两相的震荡时间为两天,以确保吸附达到平衡。
A d s o r p t i o n %t (h)图3.1 U(VI)在花岗岩上的吸附动力学;C 0,U(VI) = 2.17×10-5 mol/L ,pH = 5.65 ± 0.05,I =0.01mol/L ,T = 25o C ,m/V = 10 g/L 。
5.0x101.0x10t /q t (ht (h)图3.2 U(VI)在花岗岩上吸附动力学假二级拟合 表3.1 U(VI)在花岗岩上的吸附过程的准二级反应参数pH q e (mmol/g) K (g/mmol∙h) R 2 5.65±0.050.00114 1479.2332 0.99833.2固液比对U(VI)在花岗岩上吸附的影响图3.3给出了C 0,U(VI) = 5.21×10-5 mol/L ,pH = 5.4 ± 0.10,I = 0.01 mol/L NaCl 时U(VI)吸附百分数随固液比(m /V )的变化曲线。

第一章8.计算下列晶体的离于键与共价键的相对比例(1)NaF (2)CaO (3)ZnS解:1、查表得:X Na =0.93,X F =3.98根据鲍林公式可得NaF 中离子键比例为:21(0.93 3.98)4[1]100%90.2%e ---⨯=共价键比例为:1-90.2%=9.8% 2、同理,CaO 中离子键比例为:21(1.00 3.44)4[1]100%77.4%e---⨯=共价键比例为:1-77.4%=22.6%3、ZnS 中离子键比例为:21/4(2.581.65)[1]100%19.44%ZnS e --=-⨯=中离子键含量共价键比例为:1-19.44%=80.56%10说明结构转变的热力学条件与动力学条件的意义.说明稳态结构与亚稳态结构之间的关系。
答:结构转变的热力学条件决定转变是否可行,是结构转变的推动力,是转变的必要条件;动力学条件决定转变速度的大小,反映转变过程中阻力的大小。
稳态结构与亚稳态结构之间的关系:两种状态都是物质存在的状态,材料得到的结构是稳态或亚稳态,取决于转交过程的推动力和阻力(即热力学条件和动力学条件),阻力小时得到稳态结构,阻力很大时则得到亚稳态结构。
稳态结构能量最低,热力学上最稳定,亚稳态结构能量高,热力学上不稳定,但向稳定结构转变速度慢,能保持相对稳定甚至长期存在。
但在一定条件下,亚稳态结构向稳态结构转变。
第二章1.回答下列问题:(1)在立方晶系的晶胞内画出具有下列密勒指数的晶面和晶向:(001)与[210],(111)与[112],(110)与 [111],(132)与[123],(322)与[236](2)在立方晶系的一个晶胞中画出(111)和 (112)晶面,并写出两晶面交线的晶向指数。
(3)在立方晶系的一个晶胞中画出同时位于(101). (011)和(112)晶面上的[111]晶向。
解:1、2.有一正交点阵的 a=b, c=a/2。
某晶面在三个晶轴上的截距分别为 6个、2个和4个原子间距,求该晶面的密勒指数。
C在Ni(111)表面吸附行为的密度泛函理论研究董长青;安璐;杨勇平【期刊名称】《可再生能源》【年(卷),期】2010(028)001【摘要】应用密度泛甬理论系统地研究C在Ni(111)表面上的吸附能、吸附结构、差分电荷密度、局域态密度及Mulliken布居数,给出了覆盖度在0.167~1.0 ML 内,C的吸附特性随覆盖度变化的规律.研究表明,C的稳定吸附位为三重空位(hep,fcc),化学吸附成键的本质是C2p态和Ni3d态的耦合、杂化,使原本孤立的C 原子态耦合杂化为成键态和反键态.随着覆盖度的增加,反键态电子占据增多,成键态电子减少,平均成键电子数减少,C-Ni间的相互作用减弱,吸附能降低.【总页数】6页(P66-71)【作者】董长青;安璐;杨勇平【作者单位】华北电力大学,生物质发电成套设备国家工程实验室,北京,102206;华北电力大学,生物质发电成套设备国家工程实验室,北京,102206;华北电力大学,生物质发电成套设备国家工程实验室,北京,102206【正文语种】中文【中图分类】TK6;TK91【相关文献】1.Pt(111)和Pt3Ni(111)表面上CO催化氧化反应的密度泛函理论研究 [J], 苏海燕;李微雪;包信和2.HCN在Ni(111),Ni(100)和Ni(110)面吸附的密度泛函理论研究 [J], 辛振东;李巧玲;任君;贾秀梅3.吡啶类缓蚀剂及其在Al (111)表面吸附行为的密度泛函理论分析 [J], 吴刚;郝宁眉;廉兵杰;陈生辉;孙霜青;胡松青4.密度泛函理论研究 CO、CO +H 在 Ni(111)表面的吸附 [J], 桂岚岚;彭亮;彭导灵;顾凤龙5.C原子在Ni-Fe(111)合金表面上的吸附和扩散——密度泛函理论研究 [J], 范琛;周兴贵;成洪业;朱贻安因版权原因,仅展示原文概要,查看原文内容请购买。
第一性原理研究氧在ni(111)表面上的吸附能及功函数
近年来,随着现代科学技术的发展,金属表面催化反应的研究已经受
到了越来越多的关注。
研究人员着重研究金属表面上的吸附反应,将
其研究结果应用到催化反应中。
Ni(111)表面是一种常用的金属表面,
由于其特殊的结构,可以用来研究金属表面上的吸附反应,因此研究
Ni(111)表面上不同位置吸附原子的吸附能及其功函数的研究由来已久。
关于Ni(111)表面上氧原子的吸附能和功函数,实验上已经有大量的研究工作。
实验研究发现,氧原子的吸附能(ΔJs)和功函数(fΔJs)
均分布在结构单元上。
其中,氧原子在3个等位点位置的吸附能有所
不同,其中位于fcc位置的吸附能最高,能量为0.13eV;位于hcp位
置的吸附能和位于bridge位置的吸附能分别为0.09eV和-0.08eV。
此外,氧原子的功函数也不同,fcc位置的功函数最高,为
14.5kcal/mol,hcp位置的功函数和bridge位置的功函数分别为
7.5kcal/mol和-7.0kcal/mol。
从实验结果可以看出,氧原子在Ni(111)表面上有不同高低的吸附能和功函数,而在不同位置的吸附能和功函数也有一定差异。
这种吸附能
及功函数的分布在结构单元上,有助于更好的研究金属表面的吸附反应,以及金属表面催化反应的机制。