医疗器械临床试验与临床试验监查
- 格式:pptx
- 大小:801.10 KB
- 文档页数:30




医疗器械临床试验机构监督检查要点及判定原则
医疗器械临床试验机构监督检查要点及判定原则包括以下几个方面:
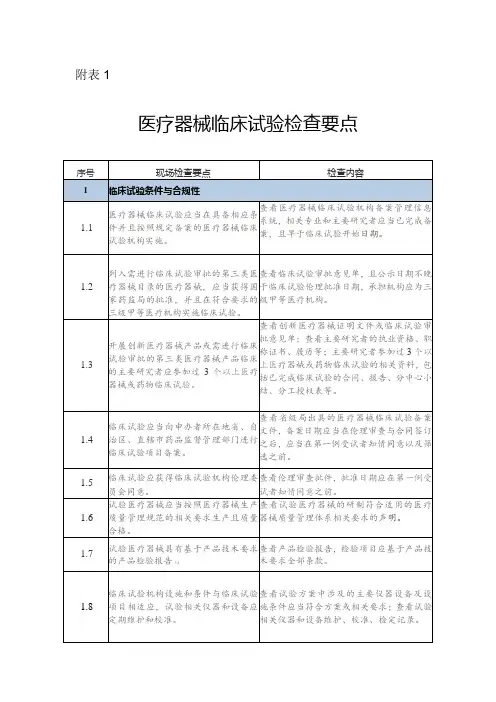
1. 试验机构管理制度:检查是否建立了科学、完整的管理制度,包括试验人员组织管理、合同管理、数据管理等方面的规定。
2. 试验人员资质和培训:核查试验人员的从业资格和相关培训情况,确保试验人员具备必要的专业知识和技能。
3. 试验设备与环境条件:检查试验机构是否配备了合适的试验设备,并保持设备的正常运行状态,同时考察试验环境是否符合相关要求。
4. 试验主体和研究方案:审查试验机构的筛选和招募试验对象的方法是否符合要求,同时检查研究方案的科学性和合理性。
5. 试验过程和数据管理:检查试验机构是否按照规定的试验程序进行试验,同时评估数据的完整性和准确性。
6. 试验结果和报告:核查试验结果的真实性和可靠性,审查试验报告的合规性和完整性。
判定原则主要包括:
1. 合规性原则:根据法律法规和监管要求,判断试验机构是否符合相关的管理规定和标准。
2. 严谨性原则:评估试验机构的试验设计、数据采集和分析,判断其科学性和方法的严谨性。
3. 可操作性原则:评估试验机构是否具备开展试验所需的设备、人员和技术条件,判断其能否按照规定的程序进行试验。
4. 有效性原则:评估试验结果的可信度和有效性,判断试验是否能够提供有关医疗器械安全性和有效性的科学依据。
以上是医疗器械临床试验机构监督检查的要点及判定原则,通过严格监督和检查,可以确保试验机构的合规性和试验结果的可信度,提高医疗器械临床试验的质量和安全性。


医疗器械临床试验与临床试验监查医疗器械临床试验是指将医疗器械应用于人体进行安全性和有效性评价的考察,是推广应用前必须进行的重要环节。
而临床试验监查则是对临床试验活动进行科学、全面、系统地监督和检查,保证临床试验活动的安全、伦理可靠以及数据的真实可信。
一、医疗器械临床试验的重要性医疗器械临床试验是确保医疗器械在实际使用中安全有效的关键环节。
通过临床试验,可以获得医疗器械在不同人群中的效果和安全性信息,为决策者提供科学的依据,指导医疗器械的投放和应用。
临床试验还可以进一步改进产品性能,提高竞争力,并为患者提供更加安全、有效的治疗方案。
二、医疗器械临床试验的内容医疗器械临床试验主要包括试验前准备、试验设计、试验实施、试验数据分析、试验报告等环节。
试验前准备包括制定试验方案、编制试验中心清单、选址和招募试验中心、制定试验合同、培训研究人员等;试验设计包括确定研究对象、制定试验方案、制定观察指标等;试验实施包括招募研究对象、进行试验操作、收集数据等;试验数据分析包括统计分析、结果解释、风险评估等;试验报告包括编写试验报告、撰写论文、申报药监部门等。
试验过程需要确保试验的科学性、规范性和伦理性。
三、临床试验监查的意义和内容临床试验监查是为了保证临床试验的安全性和伦理性,防止试验中的欺诈、误导和伦理问题的发生。
监查主要包括对试验方案和操作细节的审核、对试验中心和试验人员的审查、对试验数据的核查和监督、对试验过程的监督和评价等。
监查的目的是为了保证试验的科学性和可靠性,提高试验结果的可信度和推广价值。
四、临床试验监查的实施方式和要求临床试验监查可以通过现场监查、文件审核、数据核查、专家评审等方式进行。
监查机构可以是药监部门、独立的监测机构或者委托专业机构来进行。
监查要求监查人员具有专业知识和临床试验工作经验,遵守伦理规范和监查操作规程。
监查结果应及时反馈给试验方和监管部门,追溯问题并提出整改建议。
五、临床试验监查的挑战和应对策略临床试验监查的挑战包括试验中心的数量多、地域广、资源有限,试验人员的素质参差不齐,监查的侧重点不同,监查资源的不足等。

国家药监局关于发布医疗器械临床试验机构监督检查办法(试行)的通告文章属性•【制定机关】国家药品监督管理局•【公布日期】2024.06.14•【文号】国家药监局通告2024年第22号•【施行日期】2024.10.01•【效力等级】部门规范性文件•【时效性】尚未生效•【主题分类】药政管理正文国家药监局通告2024年第22号关于发布医疗器械临床试验机构监督检查办法(试行)的通告为进一步加强对医疗器械临床试验机构的管理,规范医疗器械临床试验机构监督检查工作,国家药监局组织制定了《医疗器械临床试验机构监督检查办法(试行)》,现予发布,自2024年10月1日起实施。
特此通告。
国家药监局2024年6月14日医疗器械临床试验机构监督检查办法(试行)第一章总则第一条为规范医疗器械临床试验机构监督检查工作,加强医疗器械临床试验管理,根据《医疗器械监督管理条例》《医疗器械注册与备案管理办法》《体外诊断试剂注册与备案管理办法》《医疗器械临床试验机构条件和备案管理办法》《医疗器械临床试验质量管理规范》等,制定本办法。
第二条药品监督管理部门对医疗器械临床试验机构(以下简称试验机构)备案及开展以医疗器械(含体外诊断试剂,下同)注册为目的的医疗器械临床试验活动,执行医疗器械临床试验质量管理规范等情况实施检查、处置等,适用本办法。
第三条国家药品监督管理局(以下简称国家局)负责制定试验机构监督检查办法,指导省级药品监督管理部门(以下简称省级局)开展试验机构监督检查,根据需要组织对试验机构进行监督检查。
国家局检查机构负责建立国家检查员库并实施检查员培训与管理,负责实施国家局组织开展的试验机构检查,推进试验机构备案管理信息化及监督检查工作信息化建设;对省级检查机构质量管理体系进行评估,对各省检查工作进行技术指导。
第四条省级局负责本行政区域内试验机构的监督检查以及国家局交办的有关事项办理,建立试验机构监督检查工作制度和机制,配备与本省试验机构检查工作相匹配的省级检查员队伍;推进监督检查工作信息化建设;组织对本行政区域内试验机构开展日常监督检查、有因检查和其他检查等,监督试验机构持续符合法定要求;对本行政区域内试验机构涉嫌违法违规行为依法进行处置。


