有机催化 α, β-不饱和羰基化合物的
- 格式:ppt
- 大小:636.00 KB
- 文档页数:27

中南大学研究生入学考试<<有机化学>>课程考试大纲【考试大纲的目的要求】一、掌握各类有机化合物的命名方法、同分异构、化合物结构及性质、化合物重要合成方法以及他们之间的相互转化关系。
二、应用价键理论的基本概念,理解有机化合物的结构;应用分子轨道理论的基本概念解释乙烯、丁二烯、苯的结构。
三、掌握诱导效应和共轭效应,并运用其解释有机物结构和性质的关系。
四、了解过渡态理论,初步掌握碳正离子、碳负离子、碳游离基等活性中间体及其在有机反应中的应用。
五、了解亲核取代、亲电取代、亲核加成、亲电加成、消去反应、游离基反应、氧化、还原、缺电子重排反应的历程。
并能初步运用来解释相应的化学反应和合成上的应用。
六、掌握常见有机金属化合物(锂、镁)的重要反应。
七、掌握立体化学的基本知识、基本理论。
八、应用红外光谱、核磁共振谱的方法测定有机化合物的结构,并能解析简单的谱图。
九、掌握各类重要有机化合物的来源、工业制法及其主要用途。
了解碳水化合物、蛋白质、油脂、主要生物碱等天然产物的结构、性质和用途。
十、掌握有机化学实验的基本技能和原理。
【考试大纲的内容】一、基本知识1. 命名与结构式(1) 系统命名烷、烯、炔、二烯、脂环(环烷、环烯、螺环和桥环)、芳烃(取代芳烃及多核芳烃)、卤代烃(卤代烷、卤代烯、卤代炔)、醇(卤代醇、芳醇)、酚(各种取代酚)、醚、醛(脂肪不饱和醛、芳醛)、酮(芳酮、环内酮)、醌、酸(取代一元酸、二元酸、芳酸、不饱和酸)、羧酸衍生物、取代酸(羟基酸、卤代酸、羰基酸)、硝基化合物、胺(芳胺)、腈、杂环化合物(母体、音译法)、碳水化合物、氨基酸的系统命名。
(2) 了解以上各类化合物的习惯命名、简单有机化合物的衍生物命名和常见化合物的俗名。
(3) 基团的命名:伯、仲、叔、季碳原子,伯、仲、叔胺、季铵,基(烷基、烯基、炔基等)的普通命名和系统命名。
(4) 写结构式:根据命名写结构式。
(5) 同系列:根据分子式、通式找出所属系列;同系物关系(同系差)。

经典化学合成反应标准操作1。
前言 (1)2. 分子内的Heck反应 (2)2.1 生成烯基取代的反应 (2)2。
1。
1 分子内Heck反应化生成环外双键示例 (3)2。
2 形成季碳中心的反应 (4)2.2。
1 分子内不对称Heck反应示例 (5)2。
3 多烯大环的合成 (5)2.2。
1 Heck反应用于合成大环多烯示例 (6)3。
分子间的Heck 反应 (7)3。
1 常规分子间Heck反应 (7)3。
1.1 Pd(OAc)2—P(o-tol)3体系用于不饱和羧酸酯的Heck反应标准操作三 (8)3.1.2 不饱和酮的Heck反应标准操作 (9)3.1。
3 杂环芳香卤代物和不饱和羧酸酯的Heck反应标准操作一 (9)3。
1。
4 杂环芳香卤代物和不饱和羧酸酯的Heck反应标准操作二 (9)3.1。
5 芳香卤代物和不饱和羧酸的Heck反应合成反式3-芳基不饱和酸示例103。
1.6 非共轭双键Heck反应示例 (10)3.2 不对称分子间Heck反应 (11)3。
3 非常用离去基团的Heck反应(Irina P。
Beletskaya Chem。
Rev. 2000,100,3009—3066) (11)3。
3。
1 重氮盐参与的Heck反应示例 (12)3.3.2 酰氯参与的Heck反应示例 (14)1。
前言通常把在碱性条件下钯催化的芳基或乙烯基卤代物和活性烯烃之间的偶联反应称为Heck反应。
自从20世纪60年代末Heck 和Morizoki独立发现该反应以来,通过对催化剂和反应条件的不断改进使其的应用范围越来越广泛,使该反应已经成为构成C-C 键的重要反应之一。
另外,Heck反应具有很好的Trans选择性R XPd(0)Z RZX = I, Br, OTf, etcZ = H, R, Ar, CN, CO2R, OR, OAc, NHAc, etc研究表明,Heck反应的机理有一定的规律,通常认为反应共分四步:(a)氧化加成(Oxidative addition): RX (R为烯基或芳基,X=I > TfO 〉Br >〉Cl)与Pd0L2的加成,形成PdⅡ配合物中间体;(b)配位插入(Cordination—insertion):烯键插入Pd—R键的过程;(c)β—H的消除;(d)催化剂的再生:加碱催化使重新得到Pd0L2。
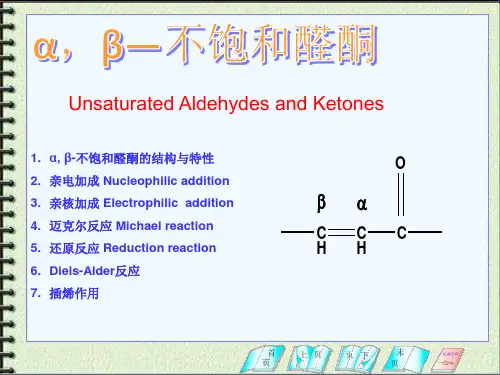

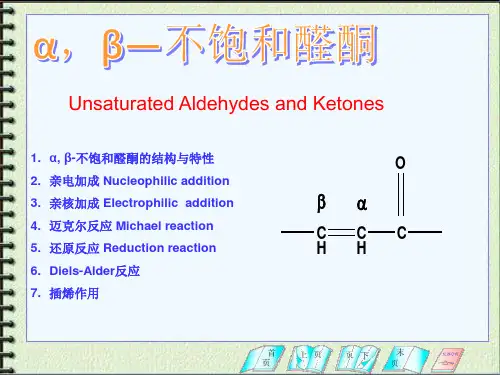


1 螺戊烷在光照条件下与氯气反应是制备氯代螺戊烷的最好方法。
Cl解释在该反应条件下,为什么氯化是制备这一化合物的如此有用的方法并写出反应历程。
解:H.该反应条件下螺戊烷氯化是自由基反应, 形成图示的平面型或近似于平面型的自由基中间体,中心碳原子为sp 2杂化, 未参与杂化的p 轨道只有一个未配对电子,垂直于三个sp 2杂化轨道,并被另一个环丙烷的弯曲键所稳定,活化能低,反应速度快,是制备该化合物有效的方法。
链引发:Cl 2链传递:Cl链终止:ClCl 2..2 解释:甲醇和2-甲基丙烯在硫酸催化下反应生成甲基叔丁基醚CH 3OC(CH 3)3(该过程与烯烃的水合过程相似)。
解:3O CH 3+- H+3下面两个反应的位置选择性不同CF3CH=CH 2CF 3CH 2CH 2ClCH3OCH=CH 2CH 3OCHClCH 3解:三氟甲基是强吸电子基,存在强的– I 效应。
生成稳定中间体碳正离子CF 3CH 2CH 2+。
连在烯键上的甲氧基存在强的+C 、弱的–I 效应,即CH 3OCH δ+=CH 2δ–,氢离子进攻 CH 2δ– ,得到中间体碳正离子CH 3OCH + CH 3也较稳定。
4解 两次亲电加成, 第一次是氢离子加到烯键上,第二次是分子内加成(碳正离子加到烯键上), 每次都生成较稳定的碳正离子。
- H +5CO 2CH 3解 +HgOAc 对烯键亲电加成后,接着经过一系列亲电加成, 再失去氢离子,得最终产物.COOCH 3OAcCOOCH 3OAcHgHg ++6Cl解 碳正离子1发生重排。
不重排的产物是1-异丙基-1-氯环己烷。
本题碳正离子重排由氢迁移造成。
ClCl++7Cl解发生碳正离子重排。
第一步得到的碳正离子已是叔碳正离子,但分子中有五元环。
重排时,碳正离子α-位环戊基的一条键带一对电子移到碳正离子上,生成六员环(1,2-迁移,碳正离子是1-位,2-位基团迁到1-位上)。



2008年第28卷有 机 化 学V ol. 28, 2008 第2期, 181~186Chinese Journal of Organic ChemistryNo. 2, 181~186* E-mail: chemjsj@ or shunjun@Received January 16, 2007; revised May 21, 2007; accepted June 7, 2007.国家自然科学基金(Nos. 20472062, 20672079)、江苏省研究生创新计划资助项目.相反, 作为应用最为广泛的有机金属试剂——格氏1 铜试剂催化下格氏试剂与环己烯酮的反应1941年Kharasch [7]报道了首例氯化亚铜催化的格氏182有 机 化 学 V ol. 28, 2008试剂与环己烯酮的反应, 得到了高区域选择性的1,4-共轭加成产物(Eq. 3), 揭开了铜试剂催化的格氏试剂与α,β-不饱和羰基化合物的1,4-共轭加成反应的研究序幕.Lippard 等[8]在1988年首次报道了用正丁基铜试剂与手性锂盐形成的新的铜络合物催化正丁基格氏试剂与环己烯酮的反应, 以14% ee , 94%的收率得到目标化合物(Eqs. 4, 5), 从而揭开了格氏试剂与α,β-不饱和羰基化合物的立体选择性反应的新篇章.在此基础上该课题组[9]又进行了深入的研究, 发现在他们的反应体系中加入HMPA 以及t -BuPh 2SiCl 能够大大提高反应的立体选择性, ee 值提高到74%, 但产率下降到57% (Eq. 6).2 S ,-N 手性配体铜络合物催化的格氏试剂的反应随后, van Koten 等[10]用S ,-N 手性配体形成的Cu(I)络合物2研究了甲基格氏试剂与开链α,β-不饱和酮的不对称1,4-共轭加成反应(Eq. 7). 他们发现, 反应的加料顺序对于反应的产率和立体选择性具有很大的影响. 将甲基格氏试剂和底物同时分别滴加至催化剂体系中, 以97%产率76% ee 得到目标化合物; 而将底物滴加至催化剂与甲基格氏试剂的混合体系中时, 产率不仅下降, 而且产物无立体选择性.1994年, 受van Koten 课题组[10]工作的启发, Pfaltz 课题组[11]用新的含有噁唑啉的S ,-N 手性配体形成的Cu(I)络合物3催化的正丁基格氏试剂与不同环烯酮的不对称共轭加成反应, 当以环庚烯酮为底物时, 反应的立体选择性最好, 以83% ee 得到目标化合物(Eq. 8).1993年, Spescha 等[12]用糖的巯基衍生物形成的铜络合物4作为催化剂研究了相同的反应, 但是ee 值只有60% (Eqs. 9, 10).1997年, Seebach 课题组[13]报道了基于TADDOL 的S ,-N 配体与铜盐的络合物5催化正丁基格氏试剂与不同环烯酮的不对称共轭加成反应, ee 值最高达到80% (Eq. 11).No. 2汪顺义等:铜络合物催化的格氏试剂不对称1,4-共轭加成反应研究进展1833 Se ,-N 手性配体铜络合物催化的格氏试剂与环状烯酮的不对称共轭加成反应2002年, Braga 课题组[14]利用双硒噁唑啉化合物6作为配体, 研究了格氏试剂与环烯酮的不对称共轭加成反应. 虽然, 产物的立体选择性较含有噁唑啉的S ,-N 手性配体催化的反应略好, 但是需要使用10 mol%催化剂, ee 值最高也只能达到85% (Eq. 12).4 P ,-N 手性配体铜络合物催化的格氏试剂的反应1997年, Sammakia 和Strangeland [15]报道了基于二茂铁噁唑啉的P ,-N 配体7参与形成的铜络合物催化的格氏试剂对α,β-不饱和酮的不对称的1,4-共轭加成反应, 这是首例利用平面手性配体参与的格氏试剂对α,β-不饱和羰基化合物的加成反应研究, ee 值最高达到92%, 但是10 mol%的催化剂的用量局限了反应的应用(Eqs. 13, 14).1999年, Tomioka 等[16]从(S )-Proline 出发, 合成了手性P ,-N 配体8, 并将其用于格氏试剂与环烯酮的不对称共轭加成反应研究, 取得了比较好的结果, ee 值最高达到92% (Eq. 15), 但是配体的用量高达32 mol%, 具有一定的局限性.2004年, Andersson 课题组[17]研究了与6相类似的P ,-N 配体9催化的正丁基格氏试剂与环己烯酮的不对称共轭加成反应, ee 值有所下降(79%) (Eq. 16).5 P ,-P 手性配体铜络合物催化的格氏试剂的反应2004~2006年, Feringa 课题组用手性二茂铁双磷配体10~12详细深入地研究了铜络合物催化下的格氏试剂与α,β-不饱和酮[18,19]、α,β-不饱和酯[20]、硫代α,β-不饱和酯[21]的不对称的1,4-共轭加成反应(Scheme 1), 取得了突破性的进展, 立体选择性得到了极大的提高(高达99% ee ) (Eqs. 17~19). 但是, 在相类似的条件下, 对于具有一定位阻的格氏试剂, 反应的转化率跟立体选择性(<10% ee )不是很理想. 在优化的条件下, 甲基格氏试剂与α,β-不饱和酯的不对称1,4-共轭加成反应的立体选择性很高(98% ee ), 但是转化率太低, 实际应用性降低. 于是, 他们改用活性更高的硫代α,β-不饱和酯代替原先的底物, 提高了反应的转化率, 而仍保持很高的立体选择性.184有 机 化 学 V ol. 28, 2008Scheme 12006年, 该课题组[22]在实验的基础上, 给出了铜络合物催化下格氏试剂与α,β-不饱和羰基化合物的1,4-共轭加成反应可能的反应机理(Scheme 2).Scheme 2我们课题组与Loh 课题组[23]合作发现, 以市售的具有平面手性的联萘双磷化合物作为配体, 能够有效地催化格氏试剂与α,β-不饱和酯的不对称1,4-共轭加成反应, 发现手性的Tol-BINAP 作为配体时反应的催化效果最好, 配体的用量只有1.5 mol% (Eq. 20).在研究中, 用(R )-Tol-BINAP 与碘化亚铜生成的碘桥联的铜的二聚络合物16(图1)作为反应的催化剂(1.0 mol%), 产物的立体选择性不是很好. 当我们增加配体与碘化亚铜比例时, 产物的立体选择性得到了提高, 物质的量比为1∶1.5时为最佳配比(图2).图1 铜络合物16的分子结构透视图Figure 1 The X-ray structure of copper dimer complex16图2 产物ee 值与(R )-Tol-BINAP 和CuI 物质的量的比的关系 Figure 2 The relationship of ee with the molar ratio of (R )-Tol-BINAP ∶CuI而对于具有一定位阻的格氏试剂, 在我们的反应体系下, 立体选择性跟转化率得到了较大的提高(Eqs. 21, 22).我们通过改变配体手性或者改变底物的几何构型能够得到相应的手性化合物, 具体关系如图3.通过对比我们发现, 反应温度对于不同的底物、不同的手性配体具有一定的影响. 在研究甲基格氏试剂No. 2汪顺义等:铜络合物催化的格氏试剂不对称1,4-共轭加成反应研究进展185α,β-不饱和酯的不对称1,4-共轭加成反应的实验过程中, 我们幸运地发现, 当适当地升高反应体系的温度, 能够大大提高反应的转化率, 适当地增加催化剂与配体的用量后, 而不影响产物的立体选择性(Eq. 23). 从而不需要使用硫代α,β-不饱和酯来代替α,β-不饱和酯与甲基格氏试剂反应. 此反应正在应用于天然产物的全合成中.6 卡宾配体铜络合物催化的格氏试剂的反应2006年, Alexakisl 课题组[24]研究了卡宾作为配体催 化的格氏试剂与3-取代的环己烯酮以及3,5,5-三取代的环己烯酮的不对称1,4-共轭加成反应, ee 值高达96% (Eq. 24).7 结论和展望近三年来, 铜络合物催化的格氏试剂1,4-共轭加成反应得到了迅速发展, 取得了一定的进展, 但是也有一些问题急待解决: 如反应活性较差立体位阻较大的格氏试剂参与的1,4-共轭加成反应. 发展新的手性配体用于铜络合物催化的格氏试剂1,4-共轭加成反应将是一个新的研究热点. 它在天然产物的合成中将会发挥越来越大的作用.图3 底物配体产物之间的构效关系 Figure 3 The configuration relationshipReferences1 (a) Alexakis, A.; Benhaim, C. Eur . J . Org . Chem . 2002,3221.(b) Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis , Tetrahedron Organic Chemistry , Series 9, Per-gamon, Oxford, 1992.(c) Rossiter, B. E.; Swingle, N. M. Chem . Rev . 1992, 92, 771.2 Review see: Li, C.; Liu, Q.; Nie, X. Chemistry 2002, 1 (inChinese).(李纯纬, 刘庆彬, 聂新永, 化学通报, 2002, 1.)3 (a) Feringa, B. L.; De Vries, H. M. Adv. Catal. Processes , 1(Asymmetric Chemical Transformations ), 1995, p. 151. (b) Krause, N. Angew . Chem ., Int . Ed . 1998, 37, 283. (c) Tomioka, K.; Nagaoka, Y. In Comprehensive Asymmet-ric Catalysis , Vol. 3, Eds.: Jacobsen, E. N.; Pfaltz, A.; Ya-mamoto, H., Springer-Verlag, New York, 1999, pp. 1105~1120.(d) Sibi, M. P.; Manyem, S. Tetrahedron 2000, 56, 8033.186有机化学V ol. 28, 2008(e) Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171.4 (a) Feringa, B. L.; Naasz, R.; Imbos, R.; Arnold, L. A. InModern Organocopper Chemistry, Ed.: Krause, N., Wiley- VCH: Weinheim, Germany, 2002, pp. 224~258.(b) de Vries, A. H. M.; Meetsma, A.; Feringa, B. L. Angew.Chem., Int. Ed. Engl. 1996, 35, 2374.(c) Feringa, B. L.; Pineschi, M.; Arnold, L. A.; Imbos, R.;de Vries, A. H. M. Angew. Chem., Int. Ed. Engl. 1997, 36, 2620.5 (a) Mizutani, H.; Degrado, S. J.; Hoveyda, A. H. J. Am.Chem. Soc. 2002, 124, 779.(b) Alexakis, A.; Benhaim, C. Eur. J. Org. Chem. 2002,3221 and references therein.6 (a) Frase, P. K.; Woodward, S. Chem. Eur. J. 2003, 9, 776.(b) Hayashi, T.; Ueyama, K.; Tokunaga, N.; Yoshida, K. J.Am. Chem. Soc. 2003, 125, 11508.(c) Alexakis, A.; Albrow, V.; Biswas, K.; d'Augustin, M.;Prieto, O.; Woodward, S. Chem. Commun. 2005, 2843.7 Kharasch, M. S.; Tawney, P. O. J. Am. Chem. Soc. 1941,63, 2308.8 Villacorta, G. M.; Rao, C. P.; Lippard, S. J. J. Am. Chem.Soc. 1988, 110, 3175.9 Ahn, K.-H.; Klassen, R. B.; Lippard, S. J. Organometallics1990, 9, 3178.10 Knotter, D. M.; Van Maanen, H. L.; Grove, D. M.; Spek, A.L.; Van Koten, G. Inorg. Chem. 1991, 30, 3309.11 (a) Zhou, Q.; Pfaltz, A. Tetrahedron Lett. 1993, 34, 7725.(b) Zhou, Q.; Pfaltz, A. Tetrahedron 1994, 50, 4467.12 Spescha, M.; Rihs, G. Helv. Chim. Acta1993, 76, 1219. 13 Seebach, D.; Jaeschke, G.; Pichota, A.; Audergon, L. Helv.Chim. Acta1997, 80, 2515.14 Braga, A. L.; Silva, S. J. N.; Lüdtke, D. S.; Drekener, R. L.;Silveira, C. C.; Rocha, J. B. T.; Wessjohann, L. A. Tetrahe-dron Lett. 2002, 43, 7329.15 Strangeland, E. L.; Sammakia, T. Tetrahedron1997, 53,16503.16 Kanai, M.; Nakagawa, Y.; Tomioka, K. Tetrahedron1999,55, 3831.17 Modin, S. A.; Pinho, P.; Andersson, P. G. Adv. Synth. Catal.2004, 346, 549.18 Feringa, B. L.; Badorrey, R.; Peña, D.; Harutyunyan, S. R.;Minnaard, A. J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5834.19 López, F.; Harutyunyan, S. R.; Minnaard, A. J.; Feringa, B.L. J. Am. Chem. Soc. 2004, 126, 12784.20 López, F.; Harutyunyan, S. R.; Minnaard, A. J.; Feringa, B.L. Angew. Chem., Int. Ed. 2005, 44, 2752.21 Des Mazery, R.; Pullez, M.; López, F.; Harutyunyan, S. R.;Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc. 2005, 127, 9966.22 Harutyunyan, S. R.; López, F.; Browne, W. R.; Correa, A.;Peña, D.; Badorrey, R.; Meetsma, A.; Minnaard, A. J.;Feringa, B. L. J. Am. Chem. Soc. 2006, 128, 9103.23 Wang, S.-Y.; Ji, S.-J.; Loh, T.-P. J. Am. Chem. Soc. 2007,129, 276.24 Martin, D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit,M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416.(Y0701163 QIN, X. Q.; LING, J.)。

Mannich反应:具有活性H的化合物与甲醛胺进行缩合,生成胺甲基衍生物的反应,亦称α-氨烷基化反应
Bcekmann重排:(亲核)肟类化合物在酸性催化剂作用下,烃基向氮原子迁移,生成取代酰胺的反应Hofmann重排:(亲核)未取代的酰胺与次溴酸钠作用,生成比反应物少一个碳原子的伯胺的反应Reformatsky反应:醛或酮与α-卤代酸酯在金属锌粉存在下缩合而得β-羟基酸酯脱水为α,β-不饱和酸酯的反应
傅克烷基化:卤代烃与芳香族化合物在Lewis酸催化下反应,在苯环上引入烃基的反应称为傅克烷基化傅克酰基化:羧酸及羧酸衍生物在质子酸或Lewis酸的催化下,对芳烃进行亲电取代成芳酮
Aldol缩合:含α活性氢的醛或酮,在酸或碱的催化下发生自身缩合,或与另一分子的醛或酮发生缩合生成β羟基醛或酮类化合物,但该类化合物不稳定,易发生消除α,β不饱和醛酮
Claisen反应:羧酸酯与另一分子具有α活泼氢的酯进行缩合得到β酮酸酯的反应又成为Claisen缩合Favorskii重排:α卤代酮和烷氧负离子作用,发生重排得到酯的反应
Michael反应:活性亚甲基化合物和α,β不饱和羰基化合物在碱性催化剂存在下发生加成缩合生成β羧烷基化合物的反应
Knoevenagel反应:具有活性亚甲基的化合物在弱碱的催化下,与醛酮发生的失水缩合反应
Darzens反应:醛或酮与α-卤代酸酯在碱催化下缩合生成α,β-环氧羧酸酯的反应
Friedel-Crafts反应:在氯化铝的催化下,卤代烃及酰卤与芳香族化合物反应,在环上引入烃基及酰基。
几类重要的不对称反应及新型手性配体欧阳志强1 欧阳迎春2(1.南昌大学材料科学与工程学院 南昌330047 2.江西师范大学理电学院南昌330027)摘 要:不对称合成是有机合成领域的热点,本文综述了以有机配体———金属配合物为手性催化剂的不对称合成的最新进展。
关键词:不对称合成 手性催化剂 手性配体 引 言自1968年美国孟山都公司的K nowlex和德国的H omer 分别发表了手性膦配体与铑配合物组成的手性催化剂进行的不均相催化氢化以来,人们相继研究开发了一大批具有立体选择性和高催化活性的新型手性配体,本文将就这方面的最新进展作一综述。
1 C =C 双键的不对称氢化反应2,2′一二(二苯基磷)一1,1′—联萘(BI NAP )的Ru络合物还年广泛用于C =C 双缝的不对称氢化。
主要有1,1′一二取代的不含杂原子的烯的不对称氢化;α,β—不饱和和β,γ—不饱和酸的不对称氢化———这类底物的不对称氢化应用于非麻醉性消炎药萘普森和异丁基布洛芬的工业生产上,潜力极大;以及前手性烯丙基醇的不对称氢化:产物为(R )一或(S )一香茅醇,ee 值高达96%-99%,香茅醇是合成L —薄荷醇的中间体。
结构与BI NAP 相似的2,2′—二氨基—1,1′一联萘(BI 2NAM )或其衍生物配体的Rh 配合物可以以36%-95%的光学收率催化a 一酰胺基丙烯酸与H 2的加成,以及前手性烯酰胺的不对称氢化,前手性酮的不对称还原。
Perea 等对平面手性二茂铁的双膦配体、氮膦配体、硫膦配体与Rh ,Ir 或Ru 形成的催化剂对C =C 的不对称氢化进行了考察,指出Rh 催化剂效果最好。
并对其反应底物的结构,溶剂效应及反应动力学等方面进行研究。
2 C =0双键的不对称还原加氢反应。
前手性酮的不对称还原得到光学活性的仲醇,2,2′—二羟基一1,1—联萘(BI NO L )改造的LiAIH 4还原剂(BI NO L -H )用于前手性不饱和酮的还原可得100%ee的相应仲醇,立体选择性依赖于温度、底物、溶剂、配位体等。
β二羰基化合物在有机合成中的应用
β-二羰基化合物是一类在有机合成中广泛应用的重要化合物。
它们的分子结构中含有两个相邻的羰基基团(C=O)。
这种基团具有很强的反应性和多种反应途径,因此能够在多种有机合成反应中作为反应物、中间体或产物起到重要作用。
2. 将β-二羰基化合物作为不饱和化合物的前体
β-二羰基化合物可以被还原形成α-羟基醛和醛,并且能够与其他分子加成反应生成新的化合物。
这些性质使得β-二羰基化合物成为制备多种不饱和化合物的重要前体,包括环己烯酮等。
其中一个典型的应用是环己烯酮的合成,反应式如下:
此外,β-二羰基化合物也可以通过Curtius重排反应,将两个羰基分别转化为亚胺基和异氰酸酯来制备不饱和化合物。
3. Michael加成反应的反应物和中间体
Michael加成反应是合成了酰胺、肽等化合物的重要反应。
β-二羰基化合物在这个过程中可以充当反应物或者中间体。
例如,在合成β-二羰基α,β-不饱和酰胺时,β-二羰基化合物可以与亲核试剂如乙烯基硫醇发生Michael加成反应。
在制备肽时,β-二羰基化合物可以作为α-氨基酸的突触剂,来促进和聚合。
4. 将β-二羰基化合物用作还原剂
β-二羰基化合物在还原过程中可以将它的两个羰基都还原为甲基,成为β-甲氧羰基化合物(M+2)(β-MeOc)。
β-MeOc可与亲电烯烃进行Diels-Alder反应,制备多环化合物。
例如,β-二羰基化合物与苯并苯的双烯烃反应可以得到稠杂芳烃骨架。
双烯加成反应一、前言双烯加成反应是有机化学中的一种重要反应,广泛应用于制备有机化合物。
本文将从以下几个方面详细介绍双烯加成反应。
二、双烯加成反应的概念双烯加成反应是指两个不饱和化合物中的双键发生加成反应,形成新的化合物。
该反应通常需要催化剂存在,并且常常伴随着环化或者开环等过程。
三、双烯加成反应的分类1. 电子不对称性双烯加成反应:指两个不饱和化合物中一个具有电子亲和性,一个具有电子供给性,或者两者同时具备。
典型的例子包括迈克尔加成反应和迪尔斯-阿尔德反应。
2. 电荷对称性双烯加成反应:指两个不饱和化合物中都具有相同的电荷性质,即均为亲电性或均为亲核性。
典型的例子包括环氧化-开环和卡宾-卡宾偶联等。
3. 立体选择性双烯加成反应:指在两个不饱和化合物中,反应发生时需要考虑它们的立体结构,通常需要特定的催化剂存在。
典型的例子包括锁骨烯加成反应和不对称二烯加成反应等。
四、双烯加成反应的机理双烯加成反应的机理多种多样,但是通常都涉及到中间体或者过渡态的形成。
下面以迈克尔加成反应为例进行讲解。
1. 迈克尔加成反应机理迈克尔加成反应是一种电子不对称性双烯加成反应,其机理如下:(1)亲核试剂进攻:亲核试剂(如胺、硫醇等)进攻α-位上的羰基碳,形成一个负离子中间体。
(2)负离子中间体:负离子中间体与具有电子供给性的不饱和化合物发生Michael加成,形成一个新的碳-碳键,并且还原负离子中间体。
(3)脱水:在碱催化条件下进行脱水,生成α,β-不饱和羰基化合物。
五、双烯加成反应在有机合成中的应用1. 迈克尔加成反应:迈克尔加成反应广泛应用于药物合成、天然产物合成等领域,例如合成心脏病药物普萘洛尔、抗癌药物多柔比星等。
2. 迪尔斯-阿尔德反应:迪尔斯-阿尔德反应是一种重要的环化反应,可以制备出具有多种不同环结构的化合物,例如制备苯并环己烷、环戊二烯基丙酮等。
3. 环氧化-开环反应:环氧化-开环反应可以制备出具有羟基或羧基官能团的化合物,例如制备马来酸二乙酯、丁二酸二甲酯等。
Knoevenagel 缩合反应文献综述1. 摘要Knoevenagel 缩合反应是有机化学中较常见的一个反应。
本文在综合大量文献的成果基础上,简述了这一反应,分析了其可能的反应机理和影响反应进行的动力学、热力学因素,列举了此反应在有机合成方面的广泛应用,对Knoevenagel 缩合反应的研究提出了新的展望。
2. 正文2.1 反应简述Knoevenagel 缩合反应(脑文格反应;克诺维纳盖尔缩合反应;柯诺瓦诺格缩合反应;克脑文盖尔缩合反应),又称Knoevenagel 反应:含有活泼亚甲基的化合物与醛或酮在弱碱催化下,发生失水缩合生成α,β- 不饱和羰基化合物及其类似物。
图 1 Knoevenagel 缩合反应通式Z 基是吸电子基团,一般为-CHO 、-COR、-COOR、-COOH、-CN、-NO2 等基团。
两个Z 基团可以相同,也可以不同。
-NO2 基团的吸电子能力很强,有一个就足以产生活泼氢。
常用的碱性催化剂有哌啶、吡啶、喹啉和其他一级胺、二级胺等。
常用的活泼亚甲基化合物有丙二酸二乙酯、米氏酸、乙酰乙酸乙酯、硝基甲烷和丙二酸等,但事实上任何含有能被碱除去氢原子的C-H 键化合物都能发生此反应。
反应一般在苯或甲苯中进行,同时将产生的水分离出去,此法所用温度较低,产率高。
Knoevenagel 反应是对Perkin 反应的改进,将酸酐改为活泼亚甲基化合物。
由于活泼氢的存在,使得弱碱作用下,能产生足够浓度的碳负离子进行亲核加成。
弱碱的使用避免了醛酮的自身缩合,因此除芳香醛外,酮和脂肪醛均能进行反应,扩大了适用范围。
Knoevenagel 反应是制备α , β - 不饱和化合物的常用方法之一。
2.2 发现历史这个反应最早是由德国化学家亚瑟·汉斯( Arthur Hantzsch )发现的, 1885 年,他用 乙酰乙酸乙酯、苯甲醛和氨反应,发现生成了对称的缩合产物 2,6- 二甲基 -4- 苯基 -1,4- 二两年之后, Knoevenagel 又开始了对这个反应的研究,他发现,在室温或 0 ℃时, 苯甲 醛与过量乙酰乙酸乙酯在催化量的哌啶作用下, 会生成双加成物 2,4- 二乙酰基 -3- 苯基戊二 酸二乙酯。
K n o e v e n a g e l缩合反应Knoevenagel缩合反应文献综述1.摘要Knoevenagel缩合反应是有机化学中较常见的一个反应。
本文在综合大量文献的成果基础上,简述了这一反应,分析了其可能的反应机理和影响反应进行的动力学、热力学因素,列举了此反应在有机合成方面的广泛应用,对Knoevenagel缩合反应的研究提出了新的展望。
2.正文2.1反应简述Knoevenagel缩合反应(脑文格反应;克诺维纳盖尔缩合反应;柯诺瓦诺格缩合反应;克脑文盖尔缩合反应),又称Knoevenagel反应:含有活泼亚甲基的化合物与醛或酮在弱碱催化下,发生失水缩合生成α,β-不饱和羰基化合物及其类似物。
图1 Knoevenagel缩合反应通式Z 基是吸电子基团,一般为 -CHO、-COR、-COOR、-COOH、-CN、-NO等基2团。
两个 Z 基团可以相同,也可以不同。
-NO2基团的吸电子能力很强,有一个就足以产生活泼氢。
常用的碱性催化剂有哌啶、吡啶、喹啉和其他一级胺、二级胺等。
常用的活泼亚甲基化合物有丙二酸二乙酯、米氏酸、乙酰乙酸乙酯、硝基甲烷和丙二酸等,但事实上任何含有能被碱除去氢原子的 C-H 键化合物都能发生此反应。
反应一般在苯或甲苯中进行,同时将产生的水分离出去,此法所用温度较低,产率高。
Knoevenagel 反应是对Perkin反应的改进,将酸酐改为活泼亚甲基化合物。
由于活泼氢的存在,使得弱碱作用下,能产生足够浓度的碳负离子进行亲核加成。
弱碱的使用避免了醛酮的自身缩合,因此除芳香醛外,酮和脂肪醛均能进行反应,扩大了适用范围。
Knoevenagel 反应是制备α,β-不饱和化合物的常用方法之一。
2.2 发现历史这个反应最早是由德国化学家亚瑟·汉斯(Arthur Hantzsch)发现的,1885年,他用乙酰乙酸乙酯、苯甲醛和氨反应,发现生成了对称的缩合产物2,6-二甲基-4-苯基-1,4-二氢吡啶-3,5-二甲酸二乙酯,也生成了少量的 2,4-二乙酰基-3-苯基戊二酸二乙酯,这是有关 Knoevenagel 反应的最早纪录。