2_3期临床试验实施要点
- 格式:ppt
- 大小:279.50 KB
- 文档页数:52
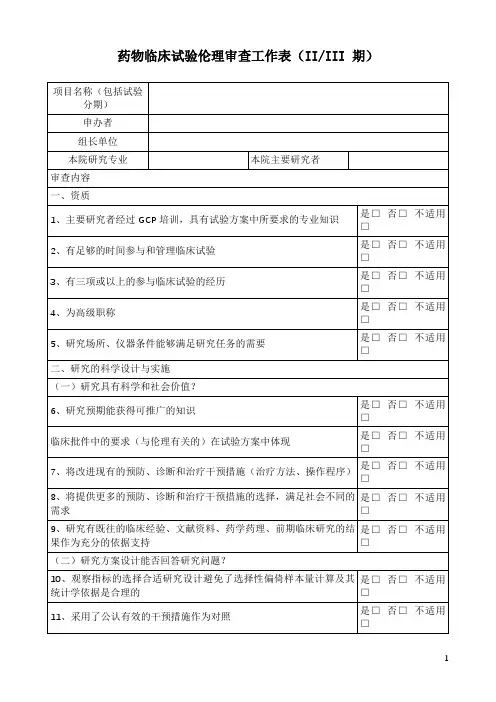

2至4期新药临床试验实施要点/原始记录内容CRF(病理报告表)质量心的严重不良事件应在获知的24小时内向机构伦理委员会医学伦理委员会申办方省和国家食品药品监督管理局总局注册司卫生行政部门报告不良事件记录事件结束随访应对所有不良事件的追踪到受试者恢复正常详细记录处理经过结果有关不良事件文件记录在原始文件所有记录均有研究者签名和日期当多个受试者发生相同不良事件而研究者手册没有应尽快报告申办方这一不良事件缩略语英文全称中文全称ADE Adverse Drug Event 药物不良事件ADR Adverse Drug Reaction 药物不良反应AE Adverse Event 不良事件AI Assistant Investigator 助理研究者BMI Body Mass Index 体质指数BE 生物等效性CI Co-investigator 合作研究者COI Coordinating Investigator 协调研究者CRA Clinical Research Associate 临床监查员(临床监察员)CRC Clinical Research Coordinator 临床研究协调者CRF Case Report Form 病历报告表CRO Contract Research Organization 合同研究组织CSA Clinical Study Application 临床研究申请CTA Clinical Trial Application 临床试验申请CTX Clinical Trial Exemption 临床试验免责CTP Clinical Trial Protocol 临床试验方案CTR Clinical Trial Report 临床试验报告DSMB Data Safety and monitoring Board数据安全及监控委员会EDC Electronic Data Capture 电子数据采集系统EDP Electronic Data Processing 电子数据处理系统FDA Food and Drug Administration 美国食品与药品管理局FIH 首次人体试验FR Final Report 总结报告GCP Good Clinical Practice 药物临床试验质量管理规范GLP Good Laboratory Practice 药物非临床试验质量管理规范GMP Good Manufacturing Practice 药品生产质量管理规范IB Investigator’s Brochure 研究者手册IC Informed Consent 知情同意ICF Informed Consent Form 知情同意书ICH International Conference on Harmonization国际协调会议IDM Independent Data Monitoring 独立数据监察IDMC Independent Data Monitoring Committee独立数据监察委员会IEC Independent Ethics Committee 独立伦理委员会IND Investigational New Drug 新药临床研究IRB Institutional Review Board 机构审查委员会IVD In Vitro Diagnostic 体外诊断IVRS Interactive Voice Response System互动语音应答系统MA Marketing Approval/Authorization上市许可证MCA Medicines Control Agency 英国药品监督局MHW Ministry of Health and Welfare 日本卫生福利部NDA New Drug Application 新药申请NEC New Drug Entity 新化学实体NIH National Institutes of Health 国家卫生研究所(美国)PI Principal Investigator 主要研究者PL Product License 产品许可证PMA Pre-market Approval (Application)上市前许可(申请)PSI Statisticians in the Pharmaceutical Industry 制药业统计学家协会QA Quality Assurance 质量保证QC Quality Control 质量控制RA Regulatory Authorities 监督管理部门SA Site Assessment 现场评估SAE Serious Adverse Event 严重不良事件SAP Statistical Analysis Plan 统计分析计划SAR Serious Adverse Reaction 严重不良反应SD Source Data/Document 原始数据/文件SD Subject Diary 受试者日记SFDA State Food and Drug Administration国家食品药品监督管理局SDV Source Data Verification 原始数据核准SEL Subject Enrollment Log 受试者入选表SI Sub-investigator 助理研究者SI Sponsor-Investigator 申办研究者SIC Subject Identification Code 受试者识别代码SOP Standard Operating Procedure 标准操作规程SPL Study Personnel List 研究人员名单SSL Subject Screening Log 受试者筛选表T&R Test and Reference Product 受试和参比试剂UAE Unexpected Adverse Event 预料外不良事件WHO World Health Organization 世界卫生组织WHO-ICDRA WHO International Conference of Drug Regulatory Authorities WHO国际药品管理当局会议Active Control 阳性对照、活性对照Audit 稽查Audit Report 稽查报告Auditor 稽查员Blank Control 空白对照Blinding/masking 盲法/设盲Case History 病历Clinical study 临床研究Clinical Trial 临床试验Clinical Trial Report 临床试验报告Compliance 依从性Coordinating Committee 协调委员会Cross-over Study 交叉研究Double Blinding 双盲Endpoint Criteria/measurement 终点指标Essential Documentation 必需文件Exclusion Criteria 排除标准Inclusion Criteria 入选表准Information Gathering 信息收集Initial Meeting 启动会议Inspection 检察/视察Institution Inspection 机构检察Investigational Product 试验药物Investigator 研究者Monitor 监查员(监察员)Monitoring 监查(监察)Monitoring Plan 监查计划(监察计划)Monitoring Report 监查报告(监察报告)Multi-center Trial 多中心试验Non-clinical Study 非临床研究Original Medical Record 原始医疗记录Outcome Assessment 结果评价Patient File 病人档案Patient History 病历Placebo 安慰剂Placebo Control 安慰剂对照Preclinical Study 临床前研究Protocol 试验方案Protocol Amendments 修正案Randomization 随机Reference Product 参比制剂Sample Size 样本量、样本大小Seriousness 严重性Severity 严重程度Single Blinding 单盲Sponsor 申办者Study Audit 研究稽查Subject 受试者Subject Enrollment 受试者入选Subject Enrollment Log 受试者入选表Subject Identification Code List 受试者识别代码表Subject Recruitment 受试者招募Study Site 研究中心Subject Screening Log 受试者筛选表System Audit 系统稽查Test Product 受试制剂Trial Initial Meeting 试验启动会议Trial Master File 试验总档案Trial Objective 试验目的Triple Blinding 三盲Wash-out 洗脱Wash-out Period 洗脱期MRSD起始剂量PAD欧盟NOAEL动物种属MTD 最大耐受剂量DLE剂量限制性名词解释双盲(double blind)是指:研究对象和研究者都不了解试验分组情况,而是由研究设计者来安排和控制全部试验。

23期临床试验实施要点解析临床试验是医学研究中至关重要的一环,其中23期临床试验作为关键步骤之一,对于新药的开发和评估具有重要意义。
本文将对23期临床试验的实施要点进行解析,旨在帮助读者深入了解该阶段的操作要求和注意事项。
一、试验设计与目标在进行23期临床试验之前,需要明确试验的设计和目标。
试验设计应该详细描述药物的使用方法、剂量、研究周期等。
同时,明确试验的主要目标,如疗效评价、安全性评估等。
设计合理的试验方案对于试验结果的准确性和可靠性至关重要。
二、样本选择与分组样本选择是23期临床试验不可或缺的一步。
合适的样本选择可以提高试验的可行性和结果的有效性。
在选择样本时,需要根据试验的目标和研究对象的特点进行合理划分,如性别、年龄分组等。
同时,对照组的选择也需要严谨考虑,确保对照组具有代表性和可比性。
三、试验过程管理23期临床试验在实施过程中需要进行全面管理,确保试验的质量和可操作性。
试验过程管理包括试验药物的供应与管理、数据采集与管理、质量控制等方面。
在试验药物的供应与管理中,要确保试验药物的合规性和安全性;在数据采集与管理中,要保证数据的准确性和完整性;在质量控制中,要进行定期的监测与分析,及时纠正偏差。
四、严格的伦理要求23期临床试验需要遵守伦理要求,确保试验过程中的研究对象的权益和安全。
在试验开始之前,必须获得合适的伦理审批,并向研究对象进行详细的知情同意。
在试验过程中,要进行适当的监测和评估,及时发现并处理任何可能的不良事件。
五、数据分析与结果解读在23期临床试验完成后,需要对试验数据进行充分的分析与解读。
数据分析包括统计学方法的应用、效果指标的计算等。
在结果解读过程中,要结合临床实际和毒副作用的评估,对试验结果进行客观和全面的评价。
六、报告撰写与提交23期临床试验结束后,需要按照相关要求编写试验报告,并提交给有关部门进行审查。
试验报告应该详细描述试验的整个过程、结果和结论,确保报告的准确性和可读性。


临床试验过程详解文档1. 临床试验概述临床试验是指在人体进行的医学研究,以评估新药、治疗方法或医疗设备的安全性和有效性。
临床试验通常分为四个阶段,分别为I、II、III和IV期。
2. 临床试验阶段详解2.1 临床试验I期(初期阶段)目的:评估新药的安全性、耐受性和药代动力学特性。
主要研究内容:- 确定药物的剂量范围和最大耐受剂量。
- 观察药物对人体产生的不良反应。
- 了解药物在人体内的吸收、分布、代谢和排泄过程。
受试者人数:通常较小,一般为20-30人。
2.2 临床试验II期(中期阶段)目的:评估新药的疗效和进一步验证安全性。
主要研究内容:- 比较药物与安慰剂的疗效差异。
- 观察药物的副作用和不良反应。
- 确定药物的最佳给药方式和剂量。
受试者人数:通常为数十到数百人。
2.3 临床试验III期(后期阶段)目的:大规模验证新药的疗效和安全性,为药物上市提供充分证据。
主要研究内容:- 比较药物与现有治疗方法或安慰剂的疗效差异。
- 观察药物的长期疗效和安全性。
- 评估药物对患者生活质量的影响。
受试者人数:通常为数百到数千人。
2.4 临床试验IV期(市场监测阶段)目的:在药物上市后对其长期疗效和安全性进行监测。
主要研究内容:- 收集药物在广泛使用条件下的疗效和安全性数据。
- 监测药物的副作用和不良反应。
- 评估药物在不同人群中的用药情况和适用性。
受试者人数:不限制,涉及广泛的患者群体。
3. 临床试验流程3.1 临床试验设计- 确定研究目的、类型(随机对照试验、队列研究等)。
- 制定研究方案和统计分析计划。
3.2 临床试验招募- 通过各种途径招募受试者。
- 对受试者进行筛选,确保其符合纳入和排除标准。
3.3 临床试验实施- 按照研究方案进行试验,包括给药、观察和数据收集。
- 确保试验过程遵循伦理原则和法律法规。
3.4 临床试验数据分析- 收集、整理和分析临床试验数据。
- 评估药物的疗效和安全性。
3.5 临床试验报告和发表- 撰写临床试验报告,包括研究背景、方法、结果和结论。



医药行业中的药物临床试验方法与实施要点药物临床试验是药物研发过程中至关重要的环节,它通过系统的科学方法评估药物的疗效和安全性,为药物是否能够上市提供重要依据。
在医药行业中,药物临床试验方法和实施要点至关重要,下面将重点介绍药物临床试验的方法和实施要点。
一、药物临床试验方法1. 临床试验的设计与分组:临床试验通常采用双盲、随机分组的方法进行,以减少偏倚和提高结果的可靠性。
双盲设计指的是患者和医生都不知道受试者所接受的是试验药物还是安慰剂,以确保结果的客观性。
而随机分组是为了避免实验结果的偏颇,将病人随机分为试验组和对照组。
2. 样本选择和纳入标准:为了保证试验结果的可靠性和推广性,样本选择非常关键。
研究者需要严格制定纳入和排除标准,确保研究对象具有相似的基本特征并符合研究目的。
3. 终点指标的选择:终点指标是衡量药物疗效的主要依据,需要根据药物的作用机制和临床疗效需求来选择。
常见的终点指标包括生存率、临床缓解率、疾病进展时间等。
4. 试验药物的剂量和给药方案:药物的剂量和给药方案是临床试验中的重要因素,需要根据药物的药代动力学特点、毒副作用和病人的特殊情况来确定。
在临床试验中,通常采用逐渐增加剂量的方法逐步寻找最佳剂量。
5. 数据收集与分析:数据的收集和分析是临床试验过程中不可忽视的环节。
研究者需要制定数据收集表,记录试验结果和观察指标,并根据预定的统计学方法进行数据的分析和解读。
二、药物临床试验的实施要点1. 倫理審查與知情同意:药物临床试验必须经过伦理委员会的审查和批准,并确保研究对象得到充分知情并签署知情同意书。
保障病人的权益和安全是药物临床试验的基本前提。
2. 试验方案和操作规范:试验方案是临床试验的指导文件,包括研究目的、研究对象纳入及排除标准、试验设计、指标选择等内容。
同时,还需要制定操作规范,确保试验过程的严谨性和一致性。
3. 研究团队的组建和培训:药物临床试验需要一个专业的研究团队进行操作和管理。

二期和三期临床实验随着科学技术的不断进步,医学领域的临床实验日益成为新药研发过程中不可或缺的重要环节。
其中,二期和三期临床实验作为关键的研究阶段,具有重要的意义和价值。
本文将着重介绍二期和三期临床实验的概念、目的和流程,并探讨其在新药开发中的重要性。
一、二期临床实验二期临床实验是在新药通过一期实验且显示潜力之后进行的一项重要研究。
其主要目的是进一步评估和验证药物的安全性和有效性,并确定最佳的给药方式、剂量和适应症范围。
在二期实验中,药物将会在适宜的病人群体中进行更广泛的测试。
通常,二期实验的样本量较一期实验要大,并且可能分为多个不同的试验分组。
在二期临床实验中,研究人员将根据特定的方案,通过严谨的设计和随机分组,确保实验数据的可靠性和有效性。
同时,他们还会仔细记录实验过程中的各个关键指标,并监测患者的治疗效果和不良反应情况。
这些数据将会在后续的数据分析中被用于判断药物的疗效和安全性。
如果二期实验获得了积极的结果,那么药物将进入下一阶段的三期临床实验。
二、三期临床实验三期临床实验是在二期实验成功并获得了积极结果之后进行的最后一阶段药物试验。
其主要目的是进一步评估药物的疗效和安全性,并为新药的注册上市提供充分的证据支持。
三期实验通常包括大规模的多中心试验,以确保实验数据的可靠性和泛化性。
在三期临床实验中,研究人员将进一步验证药物的疗效,并评估其在不同人群和不同病情下的应用效果。
同时,他们还将进一步监测药物的安全性,包括治疗期间的不良反应和严重不良事件的发生率。
三期实验的样本量通常较大,并且涉及多个试验分组,以便与现有的标准治疗进行比较。
三、二期和三期临床实验的重要性二期和三期临床实验作为新药开发过程中的关键环节,具有以下重要性:1. 确保药物的安全性:二期和三期实验能够在更广泛的患者群体中评估药物的安全性,并及时发现和记录药物的不良事件和副作用,为患者的用药安全提供保障。
2. 评估药物的有效性:二期和三期实验能够进一步验证药物的疗效,并确定最佳的给药方式、剂量和适应症范围,为患者提供更有效的治疗方案。

新药II期临床试验设计思路1 序言在I期临床试验明确了药物人体耐受性、安全性、药代动力学特点和推荐的PR2D (Recommended Phase II Dose),即可以开始启动II 期临床试验。
II期临床试验又称为探索性临床试验,是新药首次在患者身上进行、以探索有效性为目的临床试验。
由于新药临床研究费用高昂、周期较长,作为承上启下的II期临床试验设计至关重要。
申办者希望通过II期临床试验,能尽快发现很有前景的新药而不至于过早终止研究,同时又希望能尽早终止无效新药的进一步试验。
2 II期临床试验全景图I期试验侧重于新药安全性和药代动力学,而II期试验重点则转移到药效学上。
II期临床试验主要目标是初步考察新药能否在目标患者人群中建立药效学相对比较一致、药物的毒性可以接受的水平。
2.1 是什么(Who)II期临床试验主要用于评价新药在目标患者上的初步有效性和安全性。
2.2 为什么(Why)II期临床试验目的与研究药物有效性有关,其寻求回答的问题包括:①药物在I期临床确证的安全剂量范围内对某一特定适应症的有效性如何?②患者短期的新药不良反应和风险是什么?2.3 做什么(What)II期临床试验包括:①确定新药作用于目标患者的最大和最小有效剂量范围,为III期临床试验剂量提供参考;②新药产生疗效的血药浓度与药效学参数的关系,即药代动力学和药效学关系。
根据目的不同,II 期临床有时又分为IIa期和IIb期。
2.4 怎么做(How)试验顺序:一般按照IIa期、IIb期序贯实施;设计原理:II期作为探索性试验,可以采用多种设计方法,如同期对照、自身对照、开放试验、三臂试验(阳性药、安慰剂、试验药)、剂量-效应关系等的研究;受试者:目标适应症患者样本量:几十到数百人判断终点:客观缓解率等接下来以肿瘤新药的II期临床为例,介绍常见II期临床试验设计。
3 肿瘤新药的II期临床设计Ⅱ期临床试验设计方法根据有无对照组设置,分为单臂试验和随机对照试验。

医院临床试验实施计划制定要点接到新药临床研究批件后,临床药理室负责人应首先确定试验负责人及主要研究者,主要研究者一经确定,则应着手修订或制定临床试验计划。
临床试验过程是由国家食品药品监督管理局、申办者和研究者共同参与的一个过程,三者在临床试验中角色不同,职责各异,因此实施计划应就三方面进行制定。
1.申办者方面(与申办者共同制定)主要就负责该临床试验的联系人、试验药品及经费、试验进度方面做出计划。
1.1 由申办者指派联系人(姓名及联系方式),并明确该联系人在临床试验中的职能1.1.1 准备研究者手册,让研究者了解研究药品。
1.1.2 负责试验合同及临床试验方案中申办方的签字。
1.1.3 负责试验用药品的供应和收回。
1.1.4 MONTTOR 的作用。
1.1.5 严重不良反应报告的接收、上报。
1.2 试验药品及经费1.2.1 按照试验方案要求提供临床试验中所用的药品(试验药品及参比品)及对照品。
1.2.2 提供所用药品及对照品的质检报告。
1.2.3 按照合同要求及时提供试验所需经费。
1.2.4 因严重不良反应而需进行治疗的费用及相应的补偿。
1.3 对试验进度及完成时限的要求。
2.研究者方面主要以临床试验方案的组织实施及各步骤(具体参照各项的SOP)预计完成的时间、参加协作人数、预计经费支出做出详细计划。
2.1 人员组织和培训2.1.1 准备研究负责人及主要研究者简历。
2.1.2 确定其他协作人员名单及任务。
2.1.3 培训计划:主要研究者检索文献并复习、准备有关资料;对参加人员进行 GCP、试验方案、研究者手册及负责项目的 SOP等方面培训,使所有人员对研究有统一认识,能各负其责。
2.2 起草知情同意书。
2.3 伦理委员会的申请及审批准备。
2.4 受试者的召集2.4.1 确定受试者的入选人数。
2.4.2 与医学及相关人员一起确定体检项目及人选、排除、剔除标准及试验中受试者的医疗保护方案。
2.4.3 招募受试者的方式。
药物ii期临床实验药物临床实验是将新的药物应用于人体,评估其安全性和有效性的关键步骤。
其中,II期临床实验是药物研发过程中的重要环节,它对于进一步确定药物的疗效和安全性至关重要。
本文将就药物II期临床实验的目的、实施步骤和注意事项进行探讨。
一、实验目的II期临床实验的主要目的是评估药物的疗效和安全性,进一步筛选出对某种疾病有治疗效果的药物。
在I期实验中,药物的安全性已在大批健康志愿者身上进行了初步评估,因此,II期实验主要聚焦于药物的疗效。
通过对患者进行用药观察,收集病情数据,对药物的临床疗效进行评估,为进一步的III期临床实验和新药上市申请奠定基础。
二、实施步骤1. 研究设计与样本选择在II期临床实验中,研究设计需要精确而科学。
通常,实验分为对照组和实验组,对照组接受常规治疗,而实验组接受待测药物治疗。
样本选择应符合统计学要求,有代表性,并且要严格按照实验方案进行。
2. 治疗方案与用药剂量确定在II期临床实验中,针对某种特定疾病,需要明确治疗方案和用药剂量。
治疗方案包括用药的频次、用药周期等。
用药剂量需要根据前期实验数据和药代动力学评估来确定,兼顾对疾病的疗效和对人体的安全。
3. 数据收集和结果分析在实验过程中,需要准确记录患者的个人信息、病情数据和用药情况,并且及时进行数据统计和分析。
结果分析应严格按照预先设定的评价指标进行,以确定药物的疗效和安全性。
三、注意事项1. 严格遵循伦理要求在进行II期临床实验时,研究人员必须遵循医学伦理和伦理委员会的相关规定。
确保实验过程中患者的权益受到保护,药物的应用符合伦理要求。
2. 实验过程监管和控制研究人员需要对实验过程进行全程监管和控制,确保实验方案的严谨性和规范性。
定期召开数据监测委员会会议,对实验过程进行评估和监测,及时调整实验方案和用药剂量等。
3. 不良反应监测和处理在实验过程中,可能会出现患者不良反应的情况。
研究人员需要及时记录和报告不良反应,并采取相应的处理措施,包括停药、减量或调整治疗方案等。
新药临床试验过程临床试验是一种科学研究方法,用于评估新药的安全性、疗效和剂量等关键信息。
它通常分为四个阶段:I、II、III和IV。
以下是每个阶段的具体内容。
第一阶段:I期临床试验I期临床试验也被称为初步安全性研究。
该阶段旨在评估新药对健康志愿者的安全性和耐受性。
研究人员通常在健康志愿者身上进行试验,研究新药的代谢过程、适应症和最佳用药剂量等方面的信息。
这个阶段的研究通常包括小规模的人群(通常为20-100人)。
第二阶段:II期临床试验II期临床试验也被称为初步疗效研究。
该阶段旨在评估新药的疗效和安全性,并确定最佳用药剂量和用法。
研究人员通常在患有特定疾病的患者身上进行试验。
这个阶段的研究通常包括中等规模的人群(通常为100-300人)。
第三阶段:III期临床试验III期临床试验也被称为证据研究。
该阶段旨在评估新药与标准治疗或安慰剂相比的疗效和安全性。
这个阶段的研究通常包括大规模的人群(通常为1000人或更多)。
研究人员会将患者随机分组,一组接受新药,另一组接受标准治疗或安慰剂,并观察其治疗效果和安全性。
这个阶段的研究通常可以提供药物的注册申请所需的关键数据。
第四阶段:IV期临床试验IV期临床试验也被称为市场监测研究。
该阶段旨在进一步评估新药在真实临床实践中的疗效和安全性。
这个阶段的研究通常在新药上市后进行,并通过长期、大规模的观察研究患者的治疗效果和安全性。
这个阶段的研究可以进一步确认新药的疗效和安全性,并监测罕见的不良反应。
在进行临床试验的过程中,研究人员必须遵循临床实践指南和道德准则,确保试验的科学性和病人的安全。
所有参与试验的患者必须签署知情同意书,并能随时退出试验。
同时,研究人员还要定期向监管机构提交试验进展和结果的报告,并接受监管机构的审查。
总结起来,临床试验是评估新药安全性和疗效的重要过程。
从I期到IV期,研究人员通过不同的阶段逐步获取相关数据,并基于数据进行最终安全性和疗效评估。
临床实验第三期b段临床实验是医学研究中不可或缺的一环,它对于验证新药物疗效、安全性以及副作用的发现起着重要的作用。
对于处于第三期临床实验的B段,本文将从开始实验、样本选择、数据收集和效果评估四个方面进行探讨。
一、开始实验在进入第三期临床实验的B段之前,研究者必须先经过严格的合规审查和伦理伦理委员会的批准。
一旦获得批准,研究者可以开始进行实验。
首先,他们需要招募足够数量的患者参与实验,确保样本的代表性。
接着,每位参与者都需要签署知情同意书,明确知晓实验的目的、过程以及可能存在的风险。
二、样本选择为了具备代表性和可靠性,样本的选择应该经过严格的筛选。
研究者通常会确定一系列的入选标准,如年龄、性别、疾病类型等,以便筛选出符合要求的患者。
在B段中,样本数量通常较大,以增加结果的可靠性。
此外,为了避免偏见,样本的选择应当采用随机化方法,确保每个样本有相同的机会被分配到不同的实验组。
三、数据收集数据收集是临床实验的核心环节,其结果直接影响到实验的结论。
在B段中,研究者需要利用标准化的方法对患者进行观察和测量。
这包括但不限于体征观测、病情记录、实验药物的使用情况等。
为了确保数据的准确性,研究者需要严格按照操作规范进行记录,并保障数据的安全性和保密性。
四、效果评估针对新药物的疗效和副作用,研究者需要进行全面的效果评估。
评估方式可以包括但不限于临床症状观察、生化指标测量、影像学检查等。
除此之外,研究者还需要收集并分析数据,进行统计学处理和结果的解读。
评估的结果将有助于判断新药物的疗效和安全性,指导进一步的研究和应用。
总结起来,临床实验第三期B段是一项重要的工作,它需要经过严格的合规审查和伦理伦理委员会的批准,进行样本选择、数据收集和效果评估。
这一阶段的实验不仅对于验证新药物的疗效和安全性至关重要,也对于推进医学研究的进展起着积极的作用。
只有通过科学规范的临床实验,才能为患者提供更安全、有效的药物治疗。