溶出度的方法学验证
- 格式:ppt
- 大小:620.50 KB
- 文档页数:36

溶出度(释放度)检测方法建立及验证标准操作规程1.目的为保证检测工作的可靠性和可重现性,在未知样品的检测前必须对检测方法进行验证以证明所采用的检测方法适合于相应的检测要求。
2 •范围建立药品质量标准吋、药詁生产工艺变更吋、制剂组分发生变更吋、原分析方法修订时均应进行溶出度或释放度测定的方法学的验证。
3.责任人检测员、项目负责人、各级项目经理:要求系统、全面验证含量测定方法并记录整理验证数据。
4.程序4.1验证内容(以下为溶出度验证方法,释放度具体详见化学药物口服缓释制剂药学研究技术指导原则。
)溶出度系指约物从片剂或胶囊剂等固体制剂在规定的溶出介质中溶出的速度和程度,是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。
它是评价药物制剂质量的一个重要指标。
一个完整的溶出度方法验证主妾包括以下内容:(1)溶出介质及介质休积的选择;(2)溶出方法(转篮法与桨法)及其转速的选择;(3)溶出量测定方法的验证,(4)溶出度均一性试验(批内)、重现性试验(批间)等。
4.2验证方法(-)溶出度测定方法的选择溶出度测定方法的选择包括溶出介质及介质体积的选择、溶出方法(转篮法与桨法)及其转速的选择。
根据《化学药物质量标准建立的规范化过程技术指导原则》,溶出介质通常采用水、O.lmol/L盐酸溶液、缓冲液(pH值3〜8为主)。
对在上述溶出介质中均不能完全溶解的难溶性药物,可加入适量的表面活性剂,如I •二烷基硫酸钠等。
检查方法转篮法以100转/分钟为主;桨法以50转/分钟为主。
应该注意的是(1)溶出介质的体积需使药物符合漏槽条件,大杯法(第一、二法)常用体积为500〜1000ml,小杯法(第三法)常用体积为100〜250ml。
部分品种为满足在溶出量测定时药物浓度的需要,可采用低于上述限度范围的溶剂。
(2)介质、方法、转速的选择一般根据溶出曲线测定结果确定。
部分资料简单-地通过比较主药在各溶剂屮的溶解度来选择溶出介质,我们认为相同的溶剂可能会导致对不同制剂溶出行为的差异,且工艺的选择、辅料的加入能改变主约在不同溶剂屮的溶解行为,故仅考虑溶解度是不适合的;部分资料根据单点测定结果进行方法和转速选择,如盐酸左旋多巴甲酯片屮报资料中采用篮法lOOrpm和桨法75rpm比较,结果45min溶出均大于95%,故选择桨法75rpm测定溶出度, 单点测定不能很好区分不同处方和生产工艺的溶出情况,也影响溶出拐点的确定,故不合适;考虑今后大生产工艺,中报单位确定溶出度检查方法中常采用高转速或延长取样时间,取样时间与溶出曲线的拐点位置相距较远,导致溶出度测定区分能力不明显,溶出度取样时间常选择溶出曲线的拐点处后推10〜20分钟, 如果时间较长或太短,可通过适当提高或减低转速等手段重新测定溶出曲线。

溶出度检测方法建立及验证标准操作规程溶出度(释放度)检测是药物质量控制中的重要测试之一,用于评估药物的溶出性能。
溶出度测试可以确定药物在固体药物制剂中的药物溶出速率,从而判断药物的口服吸收和生物利用度。
本文将介绍溶出度检测方法的建立及验证标准操作规程。
1.仪器和试剂准备(1)溶出度仪器:常用的溶出度仪包括旋转篮法、磁力驱动法和流动池法等。
根据需求选择适合的仪器。
(2)溶出介质:根据药物特性选择适当的溶出介质,如水、缓冲液、模拟胃肠液等。
(3)试剂:如酸或硷,用于调整溶出介质的pH值。
2.样品制备(1)固体制剂:称取一定重量的固体制剂,放入溶出度仪的样品容器中,加入适量的溶出介质,封闭样品容器。
(2)液体制剂:取一定量的液体制剂,放入溶出度仪的样品容器中,封闭样品容器。
3.溶出度测试条件设定(1)旋转篮法和磁力驱动法:设定速度、旋转篮或磁力驱动子的数量等。
(2)流动池法:设定流速、温度和流动池的体积等。
4.溶出度测试操作(1)样品容器准备:根据所选的溶出度仪器选择适当的样品容器。
(2)样品装载:将样品容器放入溶出度仪器中,根据仪器要求加入预定体积的溶出介质。
(3)测试条件设定:根据所选的溶出度仪器设定相应的测试条件,如速度、温度等。
(4)样品测试:启动溶出度仪器,按照设定条件进行样品测试。
(5)结果记录:根据溶出度仪器的要求,记录样品测试结果。
5.数据处理和结果分析(1)计算溶出度:根据样品测试结果,计算出药物的溶出度或释放度。
(2)结果分析:对溶出度结果进行统计学分析,如平均值、标准偏差等。
二、方法建立1.选择合适的仪器和试剂,根据药物特性和要求选择合适的溶出度仪器和溶出介质。
2.设定溶出度测试条件,包括旋转篮法、磁力驱动法或流动池法的相关参数。
3.开展溶出度测试,根据所选的溶出度仪器和条件进行样品测试。
4.收集测试数据,根据测试结果计算药物的溶出度或释放度。
5.对测试结果进行分析和评估,根据统计学方法验证方法的准确性和可靠性。

溶出度总结一、溶出度方法的确立1、溶出方法的选择(1)篮法(B)/浆法(P),不提首选浆法或蓝法非崩解型药物(B)崩解型药物制剂中含有难以溶解、扩散的成分(P)主药或辅料为一定胶性物质(P)悬浮的制剂(B),如辅料易堵塞网孔(P,使用沉降篮)(2)小杯法:≥500ml浓度过低,较灵敏的方法仍难以进行定量测定(不能使用沉降篮,测定不能再稀释测定)。
2、溶出介质的选择(1)水:不提以水为主(pH 值无法控制,在试验过程中易发生改变,适合非pH 依赖释药)(2)人工胃液(0.01~0.1mol/L盐酸溶液, 必要时可加胃蛋白酶)(3)人工肠液(必要时可加胰蛋白酶)(4)其他缓冲液(pH值一般不超过7.6)三羟甲基氨基甲烷(Tris):缓冲范围pH7.0~9.5,低离子强度(二氟尼柳胶囊)(5)其他: 低浓度表面活性剂; 醇溶液(一般<5%)人体生理pH值在胃内为1~3.5,小肠内约为7,结肠内约为7.5(6)表面活性剂----尽量避免使用,种类和浓度需通过多个试验来验证。
•FDA 溶出度指导原则:对于难溶性药物不提倡使用有机溶剂,推荐SDS,但必须证明表面活性剂的选择和用量的合理性。
即应考察表面活性剂对药物的增溶量,以确定最少且最佳的使用浓度。
采用阳离子表面活性剂/非离子表面活性剂。
十二烷基硫酸钠(Sodium laurylsulfate SLS或SDS) —纯度;pH≦2.5聚山梨酯(吐温)20-80 (Tween20-80 ) —茴三硫片/吉非替尼片/盐酸雷洛昔芬片等溴化十六烷基三甲胺—粘度大月桂基二甲基氧化铵—替代SDS 用于胶囊剂(可以与酶配伍)3、溶出介质体积的选择使药物符合漏槽条件小规格品种一般不提倡将2粒/片投入1个溶出杯中来满足测定的灵敏度需要。
常用:大杯法:500 ~1000ml ,900ml为最普遍小杯法:100~250ml。
漏槽条件:即所用介质的体积应达到被测物质在37℃时在此介质中达到饱和溶液浓度的体积的3~10倍。

阿莫西林分散片溶出度的方法学验证【摘要】目的验证阿莫西林分散片溶出度的测定方法。
方法采用紫外分光光度法,以水900 ml为溶出介质,转速每分钟75转,在波长272 nm处测定吸光度。
结果浓度与吸光度呈良好线性,r=0.9999,低中高3种浓度的方法回收率在99.02%~100.48%之间。
结论本方法能准确、快速地测定阿莫西林分散片的溶出度。
【关键词】阿莫西林分散片;溶出度;紫外分光光度法阿莫西林为半合成广谱青霉素类药,适用于对敏感细菌所致的呼吸道感染、泌尿道感染、胃肠道感染、皮肤和软组织感染。
阿莫西林在酸性条件下稳定,胃肠道吸收率达90%。
按中国药典[1]的要求,参照有关相关文献[2],本文对阿莫西林分散片的溶出度进行方法学验证,此法可以很好控制产品质量。
1仪器与样品RC-806溶出实验仪;UV-265紫外分光光度计(狭缝2nm,比色池配对良好);AG-135电子天平;阿莫西林对照品(纯度:85.8%;来源:中检所);阿莫西林分散片(吉林显峰科技制药有限公司提供,批号20110401、20110402、20110403规格:0.25 g)及其辅料。
2试验和结果2.1测定方法取本品,照溶出度测定法(中国药典2010年版二部附录X C 第二法),以水900 ml为溶出介质,转速每分钟75转,依法操作,经30 min时,取溶液20 ml,滤过,取续滤液4.7 ml置10 ml量瓶中,加水稀释至刻度,制成每1 ml中约含130 μg的溶液,摇匀,作为供试品溶液;另取本品10片,研细,称取相当于平均片重的粉末0.4499 g置100 ml容量瓶,加水溶解并稀释至刻度,摇匀,过滤,精密量取续滤液2.6 ml置50 ml容量瓶,加水至刻度,制成每1 ml 中约含130 μg的溶液,滤过,取续滤液作为对照溶液,取上述两种溶液照紫外-可见分光光度法(附录ⅣA),在272nm的波长处分别测定吸光度,计算每片的溶出量,限度规定为标示量的80%。

溶出度方法学验证
溶出度方法学验证是一种常用的药物溶出度测试方法,用于评估固体药物制剂在特定条件下的药物释放速度。
通过该方法可以比较不同制剂的溶出行为,敏感性以及药物溶出动力学。
溶出度方法学验证通常包括以下几个步骤:
1. 准备溶出介质:选择适当的介质,如模拟胃肠液、模拟体液等,并调整其pH 和温度。
2. 准备固体药物制剂:将药物制剂加入溶出器中,例如溶出度仪器的溶出器。
3. 设定实验条件:设定溶出温度、转速、溶出介质的体积等。
4. 开始实验:启动溶出度仪器,开始记录药物溶出过程。
根据所需的时间点,取样分析溶出介质中的药物浓度。
5. 分析数据:根据取样的药物浓度数据,计算溶出度曲线、累积释放度和速率等药物溶出参数。
6. 数据解释和结果分析:根据溶出度测试的结果,评估药物制剂的溶出性能,并与其他制剂进行比较。
在溶出度方法学验证过程中,一般要注意选择合适的试验条件,如适当的转速、温度、体积等,以确保测试结果的可重复性和可比性。
同时,还要注意样品的制备、溶解度测定方法的准确性等,以提高测试的精确性。
总之,溶出度方法学验证是一种有效评估固体药物制剂溶出性能的方法,对于药物研发、质量控制和生物等效性评价具有重要意义。

溶出度总结一、溶出度方法得确立1、溶出方法得选择(1)篮法(B)/浆法(P),不提首选浆法或蓝法非崩解型药物(B)崩解型药物制剂中含有难以溶解、扩散得成分(P)主药或辅料为一定胶性物质(P)悬浮得制剂(B),如辅料易堵塞网孔(P,使用沉降篮)(2)小杯法:≥500ml浓度过低,较灵敏得方法仍难以进行定量测定(不能使用沉降篮,测定不能再稀释测定)。
2、溶出介质得选择(1)水:不提以水为主(pH 值无法控制,在试验过程中易发生改变,适合非pH 依赖释药)(2)人工胃液(0、01~0、1mol/L盐酸溶液, 必要时可加胃蛋白酶)(3)人工肠液(必要时可加胰蛋白酶)(4)其她缓冲液(pH值一般不超过7、6)三羟甲基氨基甲烷(Tris):缓冲范围pH7、0~9、5,低离子强度(二氟尼柳胶囊)(5)其她: 低浓度表面活性剂; 醇溶液(一般<5%)人体生理pH值在胃内为1~3、5,小肠内约为7,结肠内约为7、5(6)表面活性剂----尽量避免使用,种类与浓度需通过多个试验来验证。
•FDA 溶出度指导原则:对于难溶性药物不提倡使用有机溶剂,推荐SDS,但必须证明表面活性剂得选择与用量得合理性。
即应考察表面活性剂对药物得增溶量,以确定最少且最佳得使用浓度。
采用阳离子表面活性剂/非离子表面活性剂。
十二烷基硫酸钠(Sodium laurylsulfate SLS或SDS) —纯度;pH≮2、5聚山梨酯(吐温)20-80 (Tween20-80 ) —茴三硫片/吉非替尼片/盐酸雷洛昔芬片等溴化十六烷基三甲胺—粘度大月桂基二甲基氧化铵—替代SDS 用于胶囊剂(可以与酶配伍)3、溶出介质体积得选择使药物符合漏槽条件小规格品种一般不提倡将2粒/片投入1个溶出杯中来满足测定得灵敏度需要。
常用:大杯法:500 ~1000ml ,900ml为最普遍小杯法:100~250ml。
漏槽条件:即所用介质得体积应达到被测物质在37℃时在此介质中达到饱与溶液浓度得体积得3~10倍。


青蒿琥脂片溶出度测定的方法学验证目的对青蒿琥脂片的溶出度检查进行方法学验证。
方法参照《中国药典》2010年版二部附录ⅩC第二法对其溶出度进行检查[1]。
采用紫外分光光度法,在289 nm的波长处测定吸光度,进行方法学验证。
结果采用该方法测定青蒿琥脂片,分别在pH1.2、4.5、6.8、水四种溶出介质中,以浓度对吸收度进行线性回归,青蒿琥酯在10~60 μg/mL范围内浓度与吸收度呈良好的线性关系。
在以上四种溶出介质中的平均回收率和标准偏差均符合要求。
结论本法测定青蒿琥酯片溶出量方法尚可,可用于该产品的质量控制。
标签:青蒿琥脂片;溶出度测定;方法学验证青蒿琥酯是具有倍半萜结构的抗疟青蒿素的衍生物之一[1],作为新型的抗疟药,青蒿琥酯具有高效、速效、低毒等特点,而且不易产生耐受性[2]。
对原虫无性体有较强的杀灭作用,能迅速控制疟疾发作,适用于脑型疟疾及各种危重疟疾的治疗[3]。
除抗疟外,尚有治疗弓形虫病、抗肿瘤等作用[4]。
青蒿琥酯片收载于国际药典第三版以及中国药典2010年版二部,溶出度测定作为反映或模拟体内药物吸收情况的实验方法在评定口服固体制剂的质量时有重要意义[5]。
本文参考有关相关文献对青蒿琥酯片的体外溶出实验进行了方法研究[6,7],可用于该产品制定质量标准的参考。
1仪器与试药ZRS-8G智能溶出仪(天大天发科技有限公司);T90+紫外分光光度计(PG.Instruments Ltd);青蒿琥酯对照品(美国USP品牌,批号:100200-200202);青蒿琥酯原料药(来源:武汉三洹医药化工有限公司);青蒿琥酯片(来源:安徽新和成皖南药业有限公司)。
所用试剂均为分析纯,水为去离子水。
2溶出度测定的方法学验证[8]2.1 衍生化方法的确定参考中国药典2010年版二部青蒿琥酯片溶出度测定方法。
青蒿琥酯碱水解条件受到水解温度、碱液浓度、水解时间影响。
因此,需要对其测定方法进行再验证与优化。
精密称取青蒿琥酯原料药50 mg,加水溶解并制成1 000 mL,作为考察溶液。

×××片溶出度试验方法学验证1、溶出度依据国家食品药品监督管理局国家药品标准新药转正标准第二十八册×××片溶出度试验方法、《中国药典》2010年版二部溶出度测定法(附录Ⅹ C)的有关要求,并参照文献进行本品的溶出度研究。
(1)溶出介质及介质体积的选择溶出介质应根据制剂的特性选用水、0.01~0.1mol/L盐酸溶液或适宜的缓冲液(pH值一般不超过7.6),应临用新制并经脱气处理。
对于极难溶出的品种,可加适量表面活性剂,如十二烷基硫酸钠(0.5%以下),如确需使用有机溶剂,可加适量,如异丙醇、乙醇等(通常浓度在5%以下),但应有依据,并尽量选用低浓度。
溶出介质的体积一般应符合漏槽条件。
×××在水中微溶,且本品为小规格品种(规格为2.5mg),根据以上溶出介质选择的原则及已有国家标准的方法,选择已有国家标准中采用的溶出介质及体积:0.1mol/L盐酸溶液〔盐酸溶液(9→1000)〕200ml。
(2)溶出方法及其转速的选择方法的选择一般可参照下列原则:①对于非崩解型药物,宜采用转篮法。
②对于崩解型药物,在进行转篮法的整个试验过程中,确保转篮网孔的通透性尤为重要,对于处方中主药或辅料(如胶性物质)影响转篮通透性的固体制剂,一般应采用桨法。
③制剂中含有难以溶解、扩散的成分,一般应采用桨法。
④对飘浮于液面的制剂,一般应选用转篮法。
如辅料堵塞网孔则选用桨法,将供试品放入沉降篮中,并在正文中加以规定。
采用小杯法时不能使用沉降篮。
⑤小杯法主要用于在转篮法和桨法条件下,溶出液的浓度过稀,即使采用较灵敏的方法仍难以进行定量测定的品种。
转速选择的原则:在质量研究的基础上,尽量选择低转速,转篮法推荐100转/分,最低不得低于50转/分;桨法推荐50转/分,最高不超过75转/分;小杯法推荐35转/分,最高不超过50转/分。
×××在水中微溶,且本品为小规格品种(规格为2.5mg),根据以上溶出方法选择的原则、转速选择的原则及已有国家标准的方法,选择已有国家标准中采用的溶出方法及转速:小杯法,50转每分钟。

布洛芬的溶出度研究及方法学验证【摘要】布洛芬是一种非处方的非甾体抗炎药物,用于缓解疼痛、退烧和消炎。
其药物溶出度研究在制剂开发、质量控制和药物生物利用度评价中具有重要作用,方法学验证的目的是确保方法能够在实际应用中可靠和准确地产生结果,从而保证科学研究和实验结果的可信度和可重复性。
本文对布洛芬的溶出度研究及方法学验证进行综述,总结药物的溶出度研究及方法学验证的概念,并分析布洛芬的溶出度研究及方法学验证的步骤,阐述国内外相关研究进展和发展历程及重要性的同时,对其给出一些可行化建议。
【关键词】布洛芬;溶出度;方法学验证;流动池法;布洛芬是一种非处方的非甾体抗炎药物,用于缓解疼痛、退烧和消炎。
研究药物的溶出度是了解药物在不同介质中的溶解程度的重要方法之一,可以为药物的制剂设计、生产和质量控制提供重要依据[1]。
方法学验证是科学研究和实验领域中的一个重要概念,它指的是验证一个实验方法、测量方法、分析方法或程序的可靠性、准确性、精确性和适用性。
验证的结果将决定方法是否可以正式应用于相应的领域。
布洛芬作为常用药,对其进行溶出度研究及方法学验证具有极高的价值。
1概述1.1药物溶出度研究及方法学验证的概念与内容药物溶出度是指药物在给定条件下在体外模拟消化液或其他介质中溶解的速度和程度。
它是药物从固体制剂中释放出来的过程,通常在制剂开发、质量控制以及药物的生物利用度研究中具有重要意义。
药物溶出度可以通过实验测定来获得,常用的测定方法包括旋转桶法、流动池法等。
1.1布洛芬的溶出度研究的实验设置及方法学验证(1)实验目的:研究布洛芬在不同介质中的溶出度,验证药物的释放特性;(2)实验仪器和试剂:溶出度测试仪、显微镜、紫外-可见分光光度计等分析设备、布洛芬样品、模拟胃肠液(可以使用生理盐水模拟胃液,磷酸缓冲溶液模拟肠液等);(3)实验方法:常见的有旋转桶法、流动池法等;(4)实验步骤:①样品制备:准备一定量的布洛芬样品,确保样品的纯度和质量;②制备模拟体液:根据需要,制备模拟胃液和模拟肠液,以模拟不同的胃肠环境。
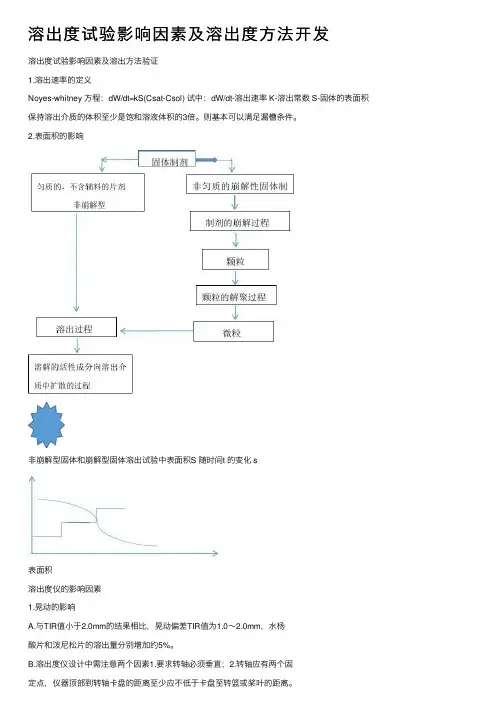
溶出度试验影响因素及溶出度⽅法开发溶出度试验影响因素及溶出⽅法验证1.溶出速率的定义Noyes-whitney ⽅程:dW/dt=kS(Csat-Csol) 试中:dW/dt-溶出速率 K-溶出常数 S-固体的表⾯积保持溶出介质的体积⾄少是饱和溶液体积的3倍。
则基本可以满⾜漏槽条件。
2.表⾯积的影响⾮崩解型固体和崩解型固体溶出试验中表⾯积S 随时间t 的变化 s表⾯积溶出度仪的影响因素1.晃动的影响A.与TIR值⼩于2.0mm的结果相⽐,晃动偏差TIR值为1.0~2.0mm,⽔杨酸⽚和泼尼松⽚的溶出量分别增加约5%。
B.溶出度仪设计中需注意两个因素1.要求转轴必须垂直;2.转轴应有两个固定点,仪器顶部到转轴卡盘的距离⾄少应不低于卡盘⾄转篮或桨叶的距离。
2.转轴的直线度a.转轴的直线度是控制晃动的关键仪器指标,应确保其直线度b.溶出度仪的涉及要求桨叶或篮体的顶端距卡盘的距离⾄少要6英⼨(15.2cm)。
3.其它搅拌装置的变动因素A.篮杆和桨杆是精密部件,使⽤时应⼩⼼,这些精密仪器在实验室的抽屉中存放时,会破坏不锈钢桨表⾯的。
引起弯曲和变性。
应有适当的⽀架供转轴存放。
B.桨叶应没有锐⾓。
尖锐部分会引起涡流⽽不是层流。
取⽤或放置篮时只能接触篮的上部边框,随着时间的增加,特别是在酸性介质时,筛⽹的孔径会有变化。
可⽤放⼤镜检,必要时需更换篮。
4.振动4.1振动的来源A实验室中能产⽣振动的仪器,包括通风橱和离⼼机,空调,风扇,离⼼机等。
B.⼈员⾛动,关门,开门。
C.早期⽔浴加热和溶出仪连在⼀起,⽬前的溶出度仪设计都采⽤外置循环泵与⽔浴连接。
4.2应将盛有溶出介质的溶出杯的各种来源的振动⽔平降低到0.1mil。
5.搅拌装置的准直度转轴的轴线与溶出杯的中⼼轴线间的偏离和倾斜对溶出介质流体动⼒学影响严重,可使溶出结果差异达到±25%。
影响结果很明显。
6.溶出杯中转轴的中⼼度7.搅拌速度规定转速⼀般不得过4%,转速的变化对溶出速率的影响⼏乎是线性的。
溶出度的方法学验证溶出度指的是单位时间内固体溶解于溶液中的量,通常以质量浓度(mg/mL)或体积浓度(mg/L)表示。
溶出度的方法学验证主要包括截短法、体外溶出试验、压片法和旋转桨法等。
截短法主要适用于缓释药物的溶出度测试。
该方法通过将靶药物的溶液倒入给定的表观容积的释放介质中,使用一定速度进行搅拌,并在不同时间点采样,测定每个时间点溶出物中药物的浓度。
然后根据浓度-时间数据绘制药物释放曲线,进而计算溶出度。
体外溶出试验也是一种常用的溶出度测试方法。
该方法通过使用自动溶出度仪来模拟体外环境,将药物制剂置于固定容器中的释放介质中,通过设定合适的搅拌速度和温度,在预定时间内采取适量的样品进行药物浓度测定。
测定结果可以用来评估制剂的溶出性能。
压片法是常用的用于固体制剂的溶出度测试方法。
该方法通过将固体制剂样品与某种溶液进行摩擦压片,使其成为固体块状,然后放入溶出介质中进行搅拌。
在一定时间内,采取适量的样品进行药物浓度测定,并根据测定结果计算溶出度。
压片法对块状制剂的溶出度测试非常适用,常用于固体制剂的质量控制。
旋转桨法用于悬浮型和非均质固体制剂的溶出度测试。
该方法使用旋转桨设备,将样品放入容器中,浸入溶出介质中并进行旋转搅拌。
在一定时间内,采取适量的样品进行药物浓度测定,并计算溶出度。
旋转桨法是一种可以模拟胃肠道溶解环境的方法,可以评估药物在溶液中的溶出速度。
除了以上常用的方法,还有其他方法可以用于溶出度的验证,如自旋诱导溶出和微量过饱和溶出等方法。
这些方法根据所需的实验条件和目的的不同,选择合适的方法来进行溶出度测试。
总结起来,溶出度的方法学验证可以通过截短法、体外溶出试验、压片法、旋转桨法等多种方法来进行。
每种方法都有其适用的药物类型和测试条件。
选用合适的方法,可以准确评估药物的溶出性能,为制药工艺控制和质量评价提供依据。
溶出度的方法学验证
溶出度是指一种物质在一定温度下在特定溶剂中的最大溶解量。
为了确定物质的溶出度,可以采用以下方法进行验证:
1. 饱和溶解度法:将一定量的物质逐渐加入溶剂中,搅拌使之充分溶解,直到不能再溶解为止。
记录此时物质的溶解量即为饱和溶解度。
2. 过饱和溶解度法:首先将一定量的物质逐渐加入溶剂中,搅拌使其充分溶解。
然后继续加入少量物质,使其超过饱和度,观察是否会出现析出物。
记录此时物质溶解的最大量即为过饱和溶解度。
3. 恒温溶解度法:在一定温度下,将一定量的物质逐渐加入溶剂中,搅拌使其充分溶解。
以一定时间间隔取样,测定取样液中物质的浓度。
当浓度不再发生显著变化时,即为恒温溶解度。
4. 温度影响法:在不同的温度下进行饱和溶解度的测定,绘制溶解度与温度之间的关系曲线。
根据曲线可以推断物质在其他温度下的溶解度。
值得注意的是,不同方法可能会在实际操作、测量精度等方面有所不同,因此在具体实验中需要选择合适的方法。
同时,需要控制实验条件(如温度、搅拌速度等)的一致性,以保证实验结果的准确性和可比性。
萘普生胶囊溶出度方法学验证方案文件编号:V-001-2013017-P河南**制药有限公司2013年09月验证方案审批表目录1、概述2、验证准备3、验证内容和方法4、验证的偏差统计分析5、验证的评价和建议6、再验证周期7、验证报告的审核8、验证报告的批准1、概述萘普生胶囊溶出度采用中国药典2010版二部附录Ⅹ C第一法进行测定,根据公司确认与验证管理规程特对此方法进行验证。
1.1目的:对萘普生胶囊溶出度检验方法进行验证,以确认本方法能对本产品的溶出度进行准确和可靠的检验,从而保证产品的质量。
1.2.范围本方案适用于萘普生胶囊溶出度检验方法的验证。
1.3依据《药品生产质量管理规范2010年版》、《确认与验证管理规程》、中国药典2010年版二部附录Ⅹ C第一法2验证准备2.1.相关的文件档案内容齐全,验证工作实施前必须有经批准现行标准文件。
2.2.验证前参加验证的人员均经过相应岗位规程及验证方案的培训,并考核合格。
参与人员培训考核情况2.3.对所用仪器仪表进行检查,确保所有仪器仪表均经过校验,并在校验期内2.4.验证时间安排验证小组制定出验证方案,经批准后实施,验证完成时间为天。
2.5验证偏差当该方案的某一部分无法实施或实际情况无法达到可接受标准时,需要进行偏差报告。
当偏差出现时,首先要进行全面调查以确定该偏差是由什么引起的,之后再确定相应的解决措施。
每个部分的偏差必须在该部分的验证工作结束之前得以解决。
也可能出现测试失败而无法解决偏差的情况,在这种情况下要对测试进行修正,且该系统的验证暂停直到方案正确修正完毕为止。
3、验证内容和方法3.1.验证内容验证的项目包括:吸收波长选择、空白干扰试验、滤膜吸附的验证、线性关系、重复性试验、精密度试验、稳定性试验、样品溶出度测定。
3.2方法描述3.2.1仪器智能溶出仪、紫外分光光度计、电子分析天平3.2.2试液溶出介质(磷酸盐缓冲液PH7.4):取磷酸二氢钠2.28g、磷酸氢二钠11.50g,加水至1000ml。
溶出度的方法学验证溶出度是指在特定条件下,其中一种物质在溶剂中溶解的最大量。
它是评价物质在特定溶剂中的溶解性质的重要指标。
溶出度的测定是药学、化学和环境科学等领域中的基础实验之一、下面将介绍几种常见的溶出度测定方法学验证。
1.旋转溶出法旋转溶出法是一种经典的溶出度测定方法。
实验中,通常采用溶出仪,将药物样品放置在旋转振荡器中,并在特定的溶剂中进行溶出试剂。
试剂的温度、搅拌速度和采样时间等条件应根据具体要求进行设定。
最后,通过测定溶出物中的药物浓度,计算得到溶出度。
2.悬浮-离心-分析法悬浮-离心-分析法是一种常用的溶出度测定方法。
实验中,药物样品通常以粉末或颗粒的形式存在。
首先将药物悬浮在溶剂中,并以设定的时间间隔进行离心。
离心后,将上清液取出进行分析,得到药物的溶出度。
离心的目的是使药物在溶液中均匀分布,提高测定的准确性。
3.透析法透析法是一种测定固体药物的离解度和溶出度的方法。
实验中,将药物样品固定于透析袋中,并将透析袋悬浮于溶剂中。
通过透析膜的作用,药物逐渐溶解并扩散到溶液中。
根据一定的时间间隔,取出溶液样品进行分析,得到药物的溶出度。
透析的时间和透析膜的选择对实验结果有一定的影响。
4.流通细胞法流通细胞法是一种体外测定溶出度的方法。
实验中,将药物样品固定于流通细胞中,并通过细胞的上下通道使溶液流通。
流通细胞法是一种连续测定溶出度的方法,因此可以评价药物溶出的时间依赖性。
通过收集溶液,测定药物浓度,可以得到溶出度曲线,进一步分析药物的释放规律。
总之,溶出度的测定方法有多种,其中旋转溶出法、悬浮-离心-分析法、透析法和流通细胞法是较常用的方法。
在进行溶出度实验时,需要根据实验目的和条件选择合适的测定方法,并注意控制实验条件的统一性和准确性,以获得可靠的溶出度数据。
此外,在实验过程中还需注意药物样品的制备、样品的取样方法以及测定药物浓度的分析方法等问题。
药物溶出度试验方法的验证概述药物溶出度试验是一项重要的药物质量控制方法,用于评估药物在特定溶媒中的溶出速度和程度。
本文将探讨药物溶出度试验方法的验证,确保试验结果准确可靠。
一、试验方法选择药物溶出度试验方法的选择应根据药物特性、制剂类型和产品要求等因素综合考虑。
常见的试验方法包括美国药典(USP)溶出度试验、欧洲药典(EP)溶出度试验和中国药典(CP)溶出度试验等。
二、验证参数的确定验证药物溶出度试验方法时,需明确以下参数:1. 单一或多个试验条件:例如,使用不同pH值、温度和搅拌速度等条件进行试验;2. 药物释放曲线的判定:通过选定特定时间点或选定时间段内累积药物释放量的百分比等方法进行判定;3. 溶出度试验仪器的准确性和重复性:对于采用自动化仪器进行试验的情况,需验证仪器的准确性和重复性,并进行适当的校准或调整。
三、实验操作步骤药物溶出度试验的实验操作步骤包括以下几个方面:1. 试剂准备:准备适当的溶媒,并根据试验方法要求进行调整;2. 试验器具准备:根据试验方法选用合适的试验仪器,并进行适当的校准和清洁;3. 试验条件设定:根据验证参数确定的试验条件进行设定,包括温度、搅拌速度等;4. 样品准备:制备样品溶液,确保样品溶液的浓度适当;5. 试验进行:将样品溶液加入试验器具中,开始试验,并记录试验时间;6. 结果分析:根据试验方法要求,进行药物释放曲线的分析和数据处理。
四、数据处理与结果分析药物溶出度试验的数据处理和结果分析要根据试验方法的要求进行。
一般来说,需要计算溶出度曲线的均值、标准差、变异系数等参数,并与设定的试验条件进行比较。
此外,还需对实验结果进行统计学分析,如方差分析等,以判断试验方法是否可靠。
五、验证报告撰写药物溶出度试验方法的验证应编写验证报告,报告内容一般包括以下几个方面:1. 验证目的和背景:明确验证的目的和背景;2. 验证参数和试验条件:列出已确定的验证参数和试验条件;3. 样品准备和试验操作:详细描述样品准备和试验操作步骤;4. 数据处理和结果分析:对试验数据进行处理和分析,并阐述结果的可靠性和合理性;5. 结论和建议:根据验证结果,给出验证结论和相关建议;6. 附录:附上试验记录和相关数据等。